
Introduction
Systemic sclerosis (SSc) is a systemic autoimmune disease characterized by vascular aberrations and tissue fibrosis. The dysfunction and damage of vascular endothelium seems to be the primary determinant in the pathogenesis. It is followed by an altered balance between injury and repair processes with abortive neoangiogenesis that results in almost complete loss of microvessels. The mechanism is complex and not fully understood [1, 2].
The hypothesis that anti-endothelial cell antibodies (AECAs) play a key role in microvascular damage in SSc seems to be increasingly convincing and experimental findings have brought new insights into their pathogenicity [3–5].
The AECAs are a heterogeneous group of autoantibodies directed against specific endothelial cells (EC) proteins and other particles adhering to their surface. These autoantibodies are suggested to play a pathogenic role in a number of autoimmune and vascular diseases characterized by endothelial damage [6]. Data concerning prevalence of AECAs in SSc patients are highly variable (22–86%) [3, 7].
Endothelial injury can be assessed in vivo by endothelium-derived biomarkers in the blood. These include circulating endothelial cells (CECs) and endothelial microparticles (EMPs) [8–12]. The CECs are mature endothelial cells that are injured and detached from the vessel wall into the bloodstream. The EMPs are microvesicles shed from the cellular membrane during ECs’ activation or apoptosis, and their release closely reflects the degree of microvascular damage. They expose specific surface markers identifying their cellular origin and are phenotypically distinct when released from either activated or apoptotic ECs [11–14].
In response to vascular injury, repair mechanisms have been shown to involve the recruitment of circulating bone marrow-derived endothelial progenitor cells (EPCs) that home into sites of insulted endothelium, differentiate into the mature endothelial phenotype and restore its integrity as well as promote neovascularization [10, 15, 16].
Aim
Based on the hypothesis that vascular damage in SSc might be associated with AECAs, we aimed to provide a combined assessment of their possible inter-relationship with the number and phenotype of EMPs and CECs. A further goal was to determine whether AECAs could affect endothelial repair capacity by counting circulating EPCs in AECA-positive versus AECA-negative SSc patients.
Material and methods
The study enrolled 47 SSc patients, referred to the Department of Dermatology, Medical University of Lublin. All patients were women and fulfilled the American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) 2013 classification criteria [17]. Forty of them were diagnosed with limited SSc (lcSSc) and 7 with diffuse SSc (dcSSc) in accordance with LeRoy et al. [18].
Demographic data was obtained from medical records. Patients with overlap syndromes, cardiovascular diseases, diabetes mellitus, hyperlipidemia, thrombosis, pregnancy, neoplastic diseases, and those with habitual cigarette smoking and alcohol drinking were excluded. Patients had a detailed laboratory and clinical assessment as described elsewhere [7, 19] and summarized in Table 1.
Table 1
Demographic and clinical characteristics of patients with systemic sclerosis enrolled in the study (n = 47)
All patients were on stable treatment for at least 6 months, including low dose prednisone (≤ 10 mg/day), vasodilators (calcium channel blockers or angiotensin-converting enzyme inhibitors) and pentoxifylline. None of the patients was on endothelin receptor blockers or treated with prostacyclin. Six patients received intravenous cyclophosphamide; however, in those patients blood samples were drawn at least 3 months after the last pulse.
The study protocol was in accordance with the Helsinki Declaration of 1975 as revised in 2000 and was approved by the institutional review board for human research at the Medical University of Lublin. All participants signed informed consent for experimentation.
AECAs identification
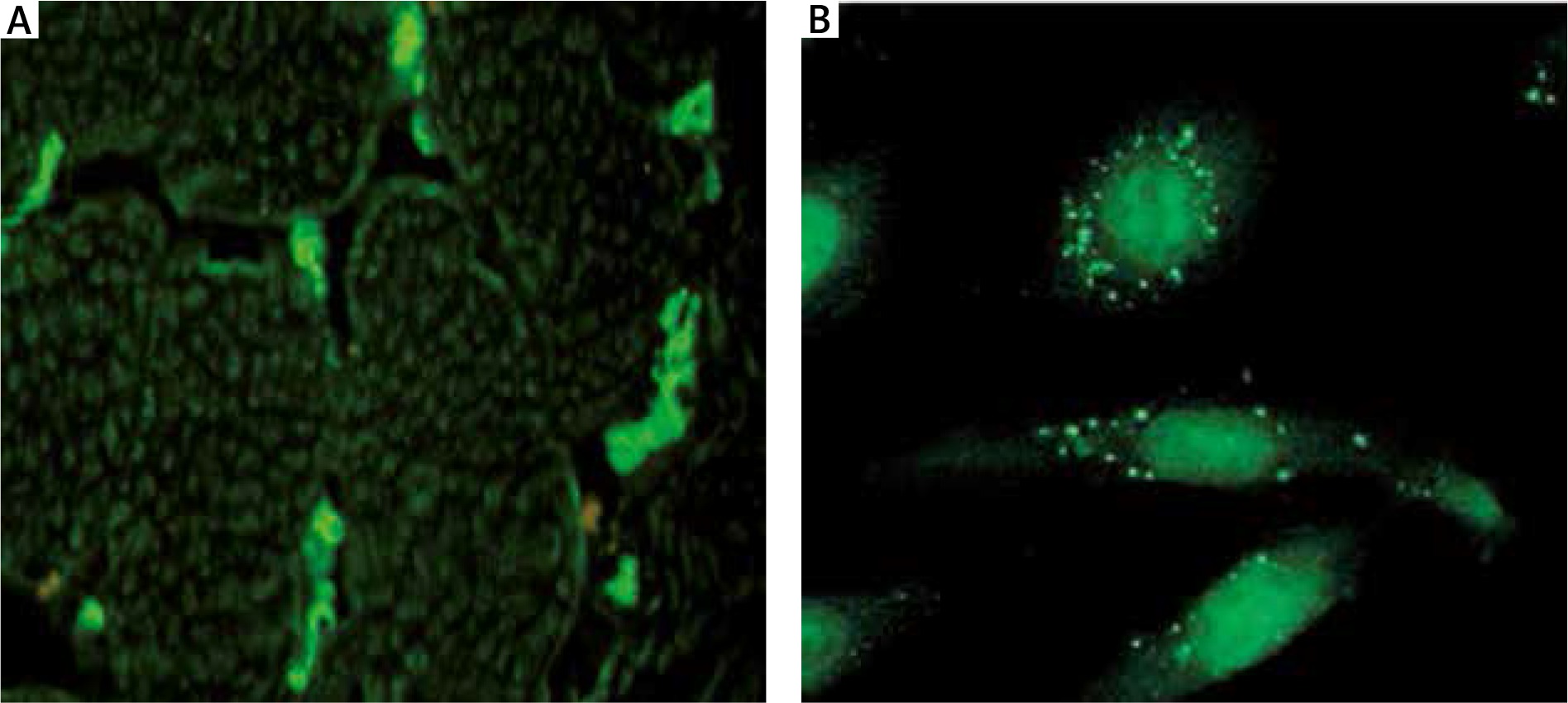
To measure the presence of AECAs, 5 ml blood samples were collected. An indirect immunofluorescence procedure with human umbilical vein endothelial cells (HUVECs) and monkey skeletal muscles was performed using TITERPLANE technique (EUROIMMUN). The procedure has been described previously [7]. Briefly, HUVECs cultures and frozen monkey skeletal muscles were incubated for 30 min with diluted 1 : 100 sera from SSc patients on biochip platforms, then rinsed with phosphate-buffered saline (PBS) Tween wash buffer and incubated with goat anti-human IgG antibodies marked with fluorescein to identify AECAs bound to the substrate. Positive reaction was assessed qualitatively using a Nikon TS100 fluorescent microscope (Figure 1).
Preparation of platelet-poor-plasma for EMPs
The EMPs were identified in platelet-poor plasma (PPP) according to the procedures described by Jimenez et al. [13] and Bernal-Mizrachi et al. [14] modified as described previously [19]. In brief, peripheral blood (5 ml) was drawn into heparinized tubes and centrifuged at 160 × g for 10 min to obtain platelet-rich plasma and then for 6 min at 1500 × g to prepare PPP. Samples of 50 µl of PPP were incubated with a combination of fluorescent monoclonal antibodies (Table 2) in the dark for 20 min at room temperature or in a refrigerator at 4°C for 45 min (anti-CD51). Staining with isotype-matched anti-IgG1 FITC/anti-IgG2a R-PE (Caltag, USA) under the same conditions was used for controls.
Table 2
Panel of antibodies applied in the identification and quantification of endothelial microparticles, circulating endothelial cells and endothelial progenitor cells by flow cytometry analysis in the present study
[i] CECs – circulating endothelial cells, EMPs – endothelial microparticles, EPCs – endothelial progenitor cells, FITC – fluorescein isothiocyanate, PE – phycoerythrin, R-PE – R-phycoerythrin, Tie2 – tyrosine kinase with Ig and EGF homology domains-2, VEGFR2 – vascular-endothelial growth factor receptor 2.
Isolation of CECs/EPCs from PBMC using the CD34 Microbead Kit
The CECs and EPCs were identified in the fresh peripheral blood according to the manufacturer’s protocol. Peripheral blood (40 ml) was drawn into heparinized tubes and processed as follows: whole blood was diluted 1 : 1 with PBS, overlaid onto an equal volume of Gradisol L (Polfa-Kutno, Poland) and centrifuged at 700 × g for 20 min. The peripheral blood mononuclear cell (PBMC) layer was carefully collected, washed twice with PBS containing 0.5% BSA plus 2 mM EDTA and counted in the Neubauer Chamber. The viability of the mononuclear cells used for the analyses was determined by the dye exclusion test and cells with viability over 95% were subjected to further analyses.
PBMC were re-suspended 1 × 108 cells in 300 µl of PBS with 0.5% BSA and 2 mM EDTA. CD34 MicroBeads (Miltenyi Biotec, Germany) and FcR blocking reagent (Miltenyi Biotec, Germany) (100 µl each for 1 × 108 cells) were added to 300 µl of PBMC suspension. Samples were incubated at 4°C for 30 min, rinsed and cells were re-suspended in PBS with 0.1% EDTA (500 µl for 1 × 108 cells).
Cells were passed through MACS separator columns (VarioMACS, Miltenyi Biotec, Germany). The CD34+ cell fraction was collected and the cell number was determined. CD34+ cells were washed with PBS + 2 mM EDTA at 700×g for 5 min at room temperature and cell suspensions were separated into 6 tubes (1 × 107 cells each) with 100 µl of PBS and labeled with different combinations of fluorescent monoclonal antibodies (5 µl of each per 100 µl of cell suspension) (Table 2). 5 µl of murine FITC-IgG1 and R-PE-IgG2a or CyChrome-IgG2a antibodies were used as a fluorescence minus control. After incubation in the dark for 30 min at 4°C, cells were washed, centrifuged and re-suspended in 500 µl of PBS for fluorocytometric analysis.
Determination of EMPs, CECs and EPCs populations by flow cytometry
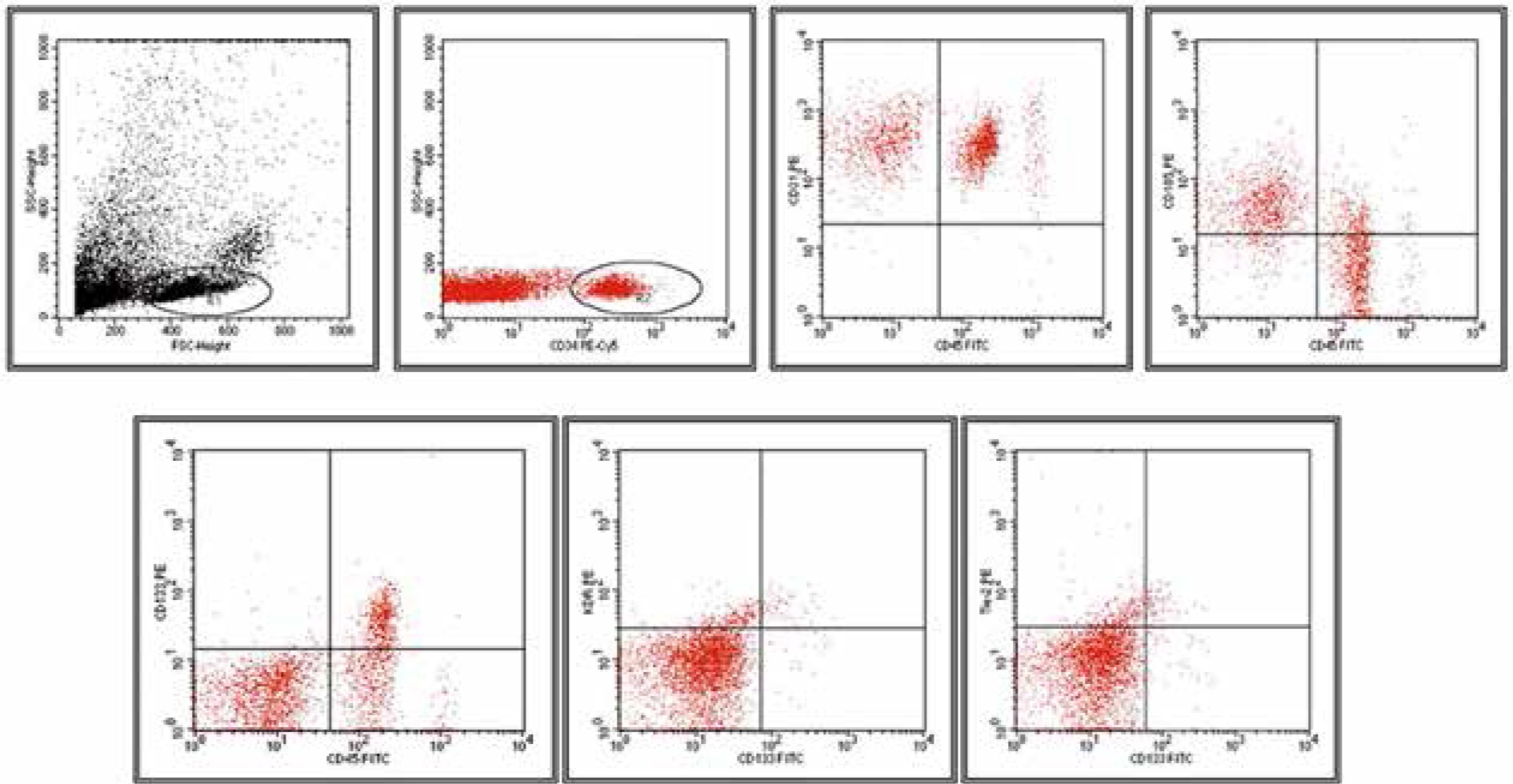
After dilution in PBS buffer, samples (1 ml) were analyzed on a flow cytometer (FACS Calibur, Becton Dickinson, USA) at a medium flow rate setting with log gain on light scatter and fluorescence. The acquisition included 500 000 events or the total volume. The gating was based on previous experience following relevant literature [10, 11, 15, 19–22]. Lymphocytes and monocytes were excluded by CD45 positivity. The EMPs were defined as particles ≤ 1.5 µm size bearing endothelial-specific antigens. Regions corresponding to microparticles were defined as described previously [19] and are presented in Figure 2. The gating strategy for identifying CD34+ CECs and EPCs is presented in Figure 3.
Figure 2
Gating strategy for endothelial microparticles (EMPs) and representative dot plots of analyzed populations
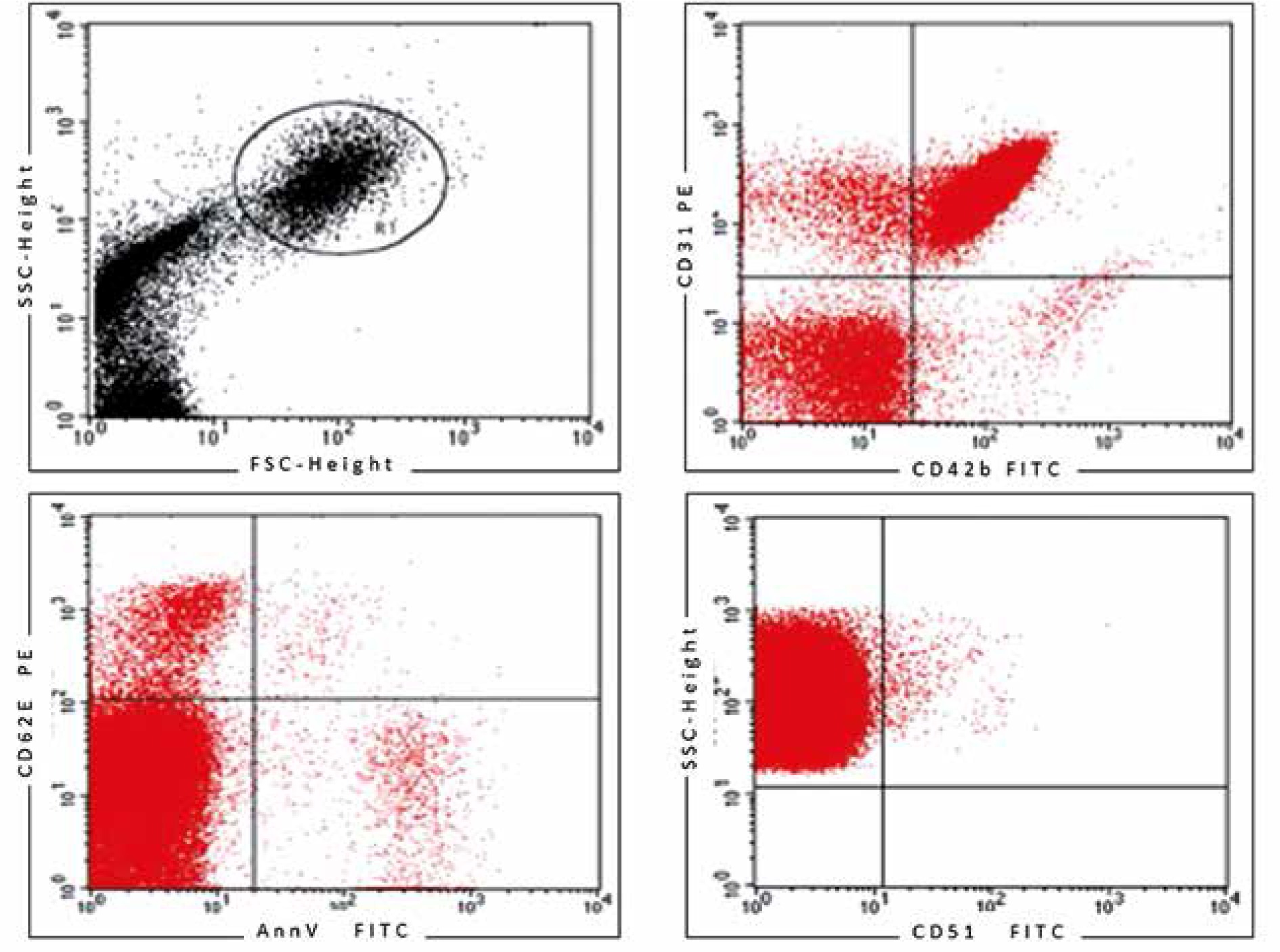
Figure 3
Gating strategy for circulating endothelial cells (CECs)/endothelial progenitor cells (EPCs) and representative dot plots of different analyzed populations
The data were evaluated with CellQuest Software (Becton Dickinson Immunocytometry Systems, USA). Values are reported as the percentage of total events for EMPs, CECs and EPCs as well as absolute values per µl of original plasma for EMPs. To convert obtained results to EMPs per µl of original plasma, the number of events was multiplied by the 1.172 dilution factor as explained elsewhere [13, 14].
We analyzed three populations of EMPs. The total population of EMPs was identified as CD31+/CD42b(–) since CD31 (PECAM-1) is present on both activated and apoptotic EMPs. The rationale of the 2-color method (CD31 and CD42b) was that significant expression of CD31 occurs on both EMPs and platelet microparticles, whereas CD42b is restricted to platelets. Microparticles released during activation were characterized as CD62E+/AnnV-, whereas EMPs from apoptotic cells were characterized as CD62E+/AnnV+ or alternatively as CD51+ [11, 13, 14]. The representative dot plots are shown in Figure 2.
For CECs and EPCs determination we created five different panels based on relevant literature and used appropriate analysis gates to analyze various phenotypes [10, 14, 20–22] (Table 2, Figure 2). According to Mancuso et al. [10] and Rafii [23] EPCs were identified within the CD34+ population based on CD133+ expression and negative for pan-leukocyte marker CD45. Alternatively EPCs were analyzed through CD34+/CD133+/VEGFR2+ and CD34+/CD133+/Tie-2+ expression. CECs of resting phenotype were deemed as CD34+/CD45-/CD31+, whereas CECs of activated phenotype were deemed as CD34+/CD45-/CD105+ [10, 24] (Table 2, Figure 3).
Statistical analysis
To evaluate the differences between the subgroups of patients the unpaired two-sample Student’s t-test was used. Statistical analysis was performed using Statistica 12.5 (StatSoft, Poland) and Graph Pad Prism 7 (Graph Pad Software, San Diego, CA, USA). The level of statistical significance was set at p < 0.05. All results are presented as mean ± standard deviation (SD).
Results
Patients were classified as either having (44.7%, n = 21) or not having (55.3%, n = 26) AECAs.
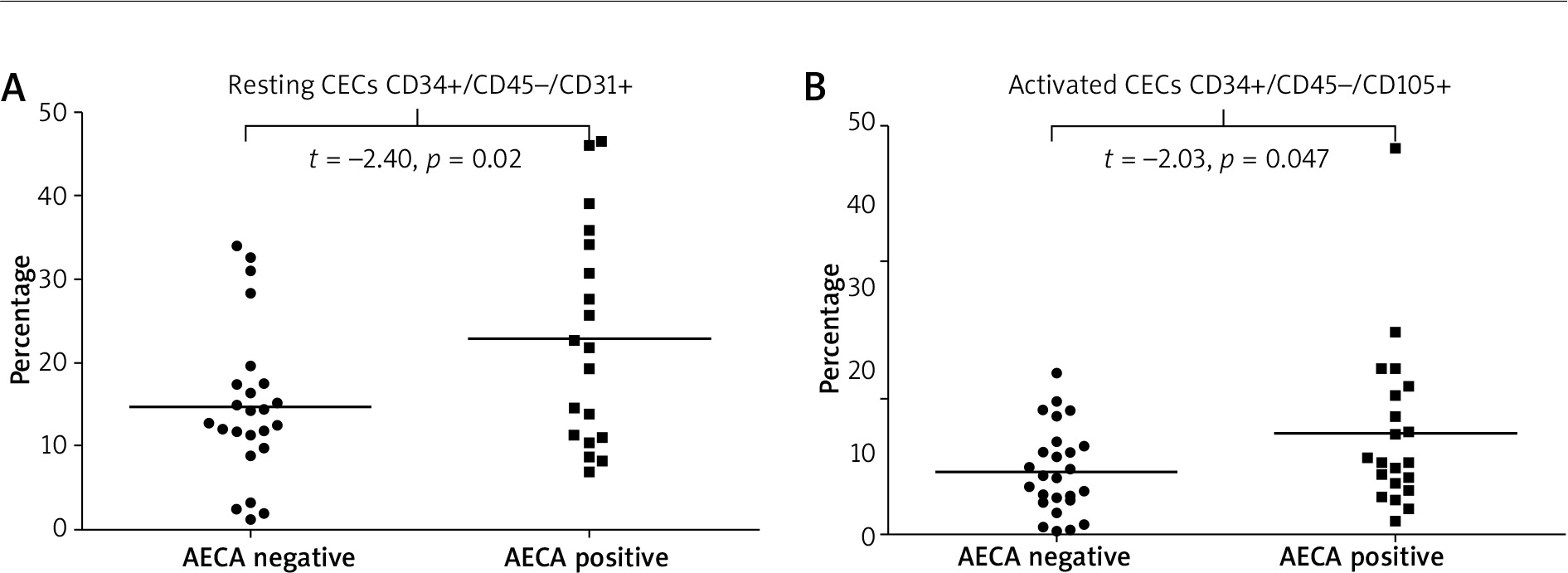
Observed associations between the presence of AECAs and endothelial markers were as follows: i) AECA-positive patients had significantly higher values of total plasma EMPs [CD31+/CD41(–)] compared to those who were negative for these autoantibodies (p = 0.037) (Figure 4 A), ii) SSc patients with AECAs had significantly higher plasma levels of apoptotic EMPs, both CD62E+/AnnV(+) and CD51(+) (p = 0.02 and p = 0.03, respectively), whereas level of activated EMPs (CD62E+/AnnV–) did not differ according to the presence or absence of AECAs (Figures 4 B–D), iii) in patients who were AECA positive we found significantly elevated total CECs CD34+/CD45 (–)/CD31+ as well as activated CD34+/CD45(–)/CD105+ CECs compared with the patient group without AECA activity (p = 0.020 and p = 0.047 respectively) (Figure 5).
Figure 4
Total, activated and apoptotic endothelial microparticles (EMPs) data for anti-endothelial cell antibody-positive and anti-endothelial cell antibody-negative systemic sclerosis patients presented as univariate scatterplots; Student’s t-test
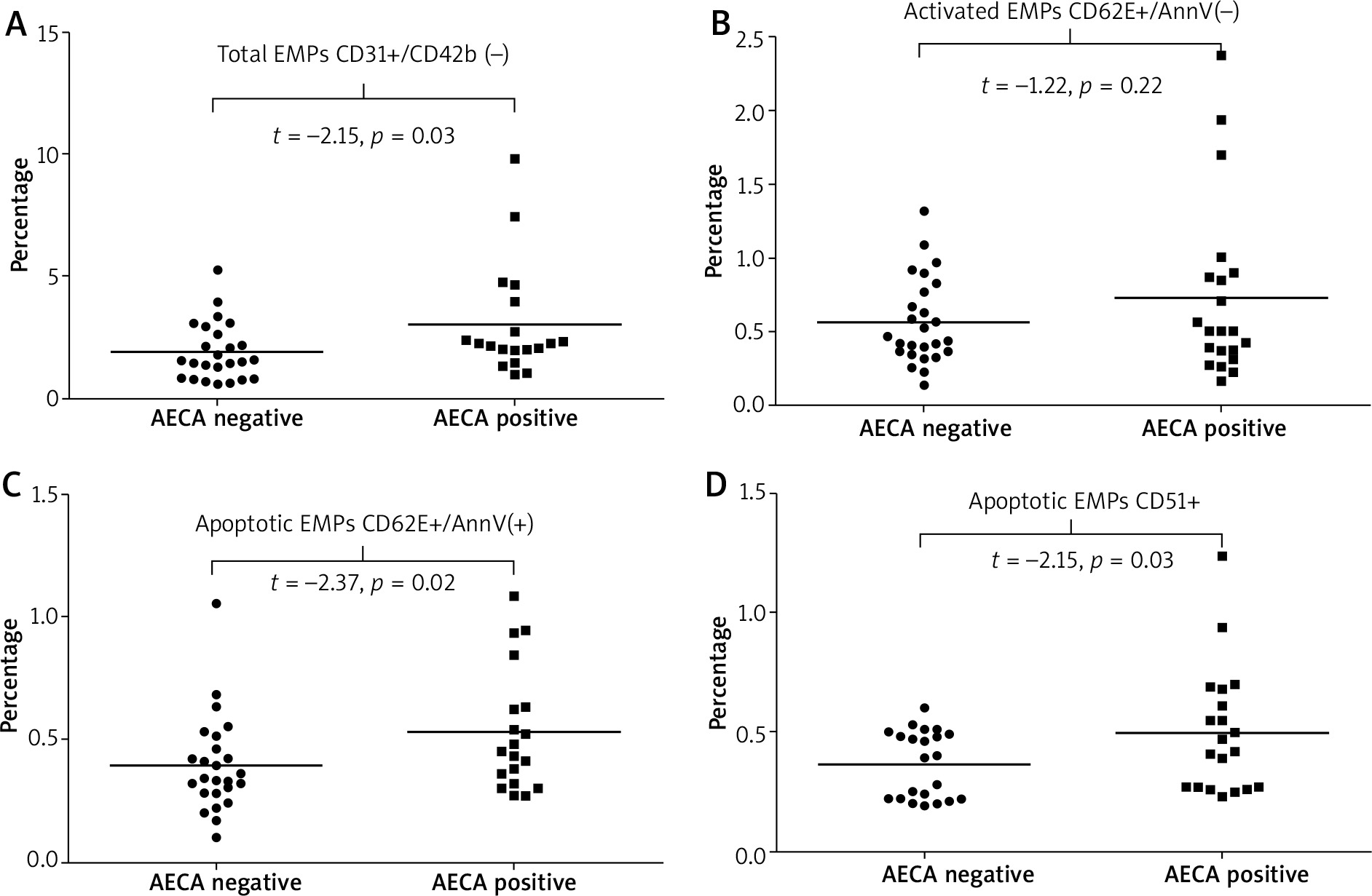
Figure 5
Resting and activated circulating endothelial cells (CECs) data for anti-endothelial cell antibody-positive and anti-endothelial cell antibody-negative systemic sclerosis patients presented as univariate scatterplots; Student’s t-test
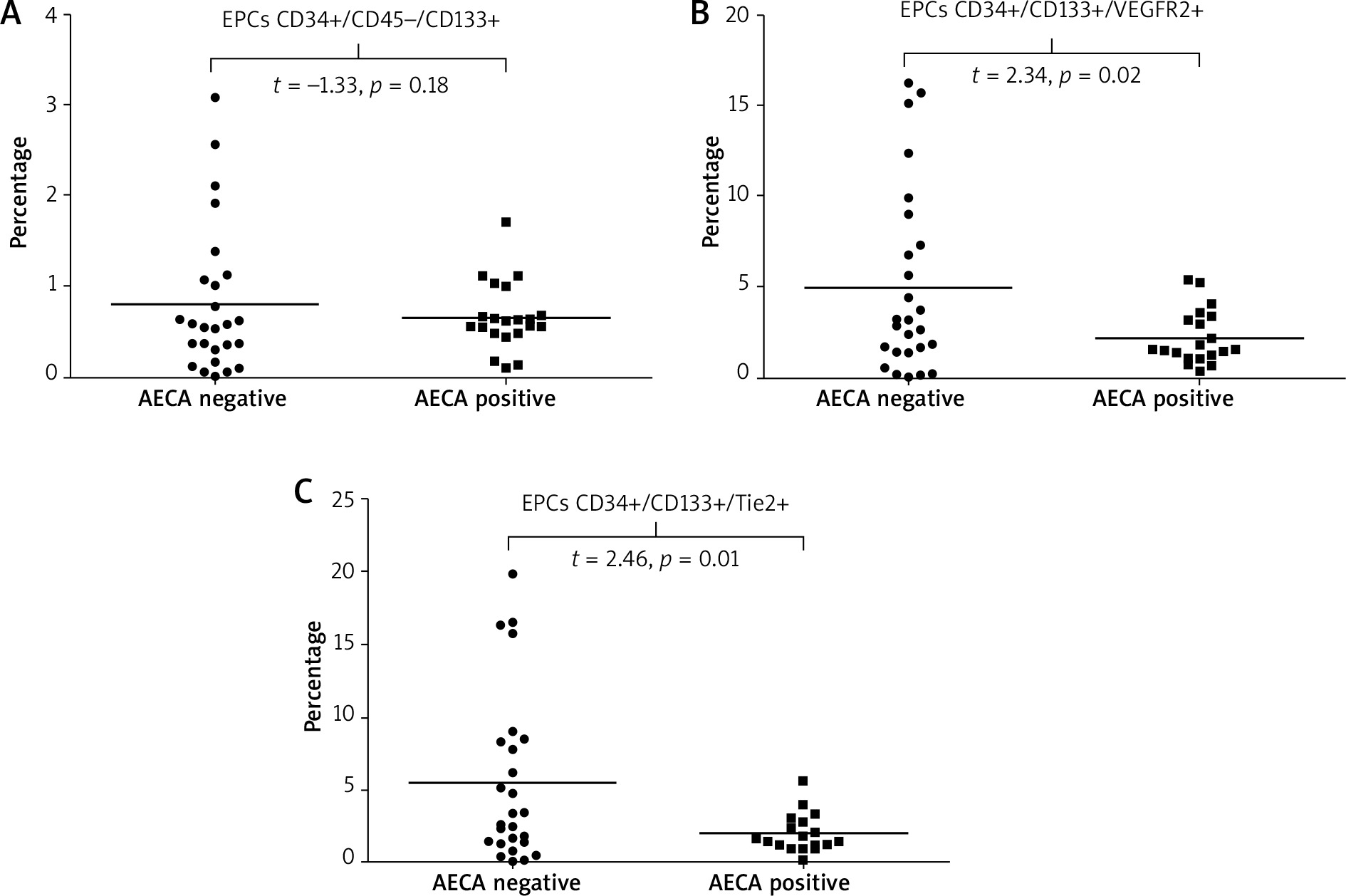
When we analyzed the relationship between AECAs and cellular markers of neovascularization, we found that: i) values of endothelial progenitors CD34+/CD45(–)/CD133+ did not differ between patients with or without AECAs (Figure 6 A), ii) patients with AECAs had a significantly lower number of EPCs expressing the receptor for VEGF, (CD34+/CD133+/VEGFR2+) as well as a significant decrease in those EPCs that express the receptor for angiopoietins (CD34+/CD133+/Tie-2+ EPCs) compared with AECA-negative subjects (p = 0.023 and p = 0.017, respectively) (Figures 6 B, C). Detailed results are summarized in Table 3.
Table 3
Results of comparison of endothelial microparticles, circulating endothelial cells and endothelial progenitors cell values between anti-endothelial cell antibody-positive and anti-endothelial cell antibody-negative systemic sclerosis patients. Student’s t-test
Discussion
The major findings of this study to be discussed are as follows: 1) in SSc patients the presence of AECAs is related to higher levels of total and activated CECs, 2) AECAs were positively associated with the number of apoptotic EMPs, but not with those of activated phenotype, and 3) endothelial progenitor cells of “angiogenic” phenotype were significantly lowered in peripheral blood of AECA-positive SSc patients.
Data from several in vitro studies suggest that in SSc AECAs mediate the endothelial cell damage and are directly linked to vascular injury, but evidence for the in vivo effects of these autoantibodies in SSc patients is still emerging [3–5, 25].
Recent findings indicate that EMPs and CECs may represent direct clinical markers of ongoing vascular disease, and thus might be helpful in assessing the extent of vascular injury in SSc [19, 20, 26]. It has been demonstrated that SSc is associated with increased levels of circulating CECs and EMPs, and EMPs reflect the progressive loss of capillaries as well as attempts of revascularization objectified by nailfold videocapillaroscopy (NVC) [19, 20].
Although there are several studies focused on either EMPs and CECs or AECAs in SSc, to our knowledge the combined assessment of the inter-relationship between these markers has not been previously reported.
In the present study we found an evident association between the presence of AECAs and significant increase in circulating total EMPs and CECs, thus giving an insight into the potential contribution of these autoantibodies to the severity of endothelial damage in SSc.
In fact, such correlation might be supported by previous reports of clinical associations between AECAs and an increased incidence of vascular manifestations, including the Raynaud phenomenon, digital ulcers and gangren, and pulmonary arterial hypertension [25, 27, 28]. It has also been found that AECA-positive patients had more severe NVC changes, in particular the late capillaroscopic pattern [25, 28, 29].
Anti-endothelial cell antibodies may exert some of their pathological effects by inducing ECs apoptosis [6, 30, 31]. In SSc, ECs apoptosis, leading to injury of the microcapillary endothelium, is thought to be the primary pathogenic event and previous in vitro studies suggest a primary role for AECAs in mediating ECs apoptosis in SSc [1, 4, 5, 30].
However, IgG from AECA-positive SSc patients has been shown to induce HUVEC activation in vitro, characterized by significantly higher expression of intracellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin (CD62E) compared to AECA-negative patients [32].
In this context, in the present study, we additionally quantified subpopulations of CECs and EMPs which may reflect these two processes, namely ECAs activation and apoptosis, to gain some insight into the mechanism of AECAs pathogenicity in SSc. Based on literature data and our previous research experience, we assigned EMPs which reflect ECs apoptosis either by the expression of CD51 or Annexin V binding capacity, whereas activated EMPs were marked by CD62E (E-selectin) expression [9, 11–13, 19]. To assess whether CECs are in an activated state, we analyzed them for the presence of the CD105 (endoglin) molecule that is expressed on the endothelial surface upon activation [10, 24].
We found AECAs to be positively associated with the number of apoptotic EMPs, either AnnV binding or CD51 positive, but not with those of activated, E-selectin positive phenotype. This observation may support the conclusion that in SSc, AECAs induce ECs apoptosis rather than their activation. It is in line with Bordron et al. [31], who reported the first evidence that AECA-positive sera from SSc patients are capable of inducing significant increase in endothelial cell apoptosis [4]. Sgonc et al. also reported Fas (CD95)-mediated ECs apoptosis after incubation of human ECs with AECAs from sera of SSc patients [5]. Adversely, some other authors reported Fas-independent apoptosis [31]. Of particular relevance to our results, Bordron et al. [31] showed that in SSc binding of AECAs to ECs was an absolute prerequisite for the externalization of phosphatidylserine (PS) and subsequent binding to annexin V that precluded apoptosis – as a result, ECs became annexin V positive as they became apoptotic [31]. Based on the literature search (PubMed, EMBASE, and Web of Science, last updated July 2018) using keywords related to either “endothelial microparticles” or “circulating endothelial cells” and “systemic sclerosis”, “scleroderma”, and “anti-endothelial cells antibodies” we have not found reports to compare with our results. In MCTD, opposite to our observations, AECAs have been found to provoke the surface expression of E-selectin (CD62E) and the activation of ECs [33]. In systemic lupus erythematosus (SLE), EMPs counts, whether total CD146+, activated CD54+ or apoptotic CD54(–), were not significantly higher for patients with AECAs and thus the authors concluded that AECAs do not seem to be the main cause of endothelial dysfunction in this population [34]. In turn, in the study of del Papa et al. [35] AECAs from SLE patients induced dose-dependent ECs activation in vitro with up-regulation of E-selectin expression. Taking into account our observations, these discrepancies may indicate different mechanisms of AECA-driven ECs injury in SSc and either SLE or MCTD; however, it needs direct comparative studies.
Intriguingly, although levels of activated EMPs (CD62E+/AnnV-) did not differ according to the presence of AECAs in our SSc group, thus testifying against AECA-driven ECs activation, we found a significant increase in activated CD105-positive CECs in those patients who were AECA positive. This might result from the fact that E-selectin is expressed exclusively on activated ECs and is cleaved from ECs that are activated but not damaged; thus the increase in activated E-selectin positive EMPs might reflect early ECs dysfunction in contrast to activated CECs, whose detection requires severe injury and sloughing of ECs [8, 11–13, 20]. Therefore, it might be suggested that in SSc presence of AECAs is unlikely associated with ECs activation, but rather with severe ECs damage resulting in their detachment, thereby potentially facilitating vascular manifestations of the disease.
Regarding activated CD105+ CECs, they are also considered a hallmark of tissues undergoing angiogenesis in vivo and are likely to contribute to new vessel formation [10, 24, 36, 37]. It has been documented that activated CECs may derive from newly formed vessels or, alternatively, represent ingress of proliferating ECs [24]. Thus, the significant increase in CD105+ CECs among our AECA-positive patients might not only reflect endothelial damage but also be a consequence of the facilitated angiogenic response induced by more severe vascular injury.
It has also been postulated that AECAs may play a pathogenic role in vascular diseases by affecting the bone marrow EPCs [38, 39]. Patients with SSc are known to have significantly fewer and functionally impaired EPCs in peripheral blood and bone marrow; however, the underlying mechanism is unclear [16, 39–41]. Thus, in the present study we additionally determined whether AECAs could affect endothelial repair capacity and neovascularization by counting circulating EPCs in AECA-positive versus AECA-negative SSc patients.
According to Rafii [23], we identified EPCs within the CD45-negative non-hematopoietic CD34+ cell fraction by surface expression of CD133, a stem cell marker that is expressed on progenitors but not on mature ECs [16, 42–44]. Alternatively, according to Asahara et al. [22] and recent EULAR Scleroderma Trials and Research (EUSTAR) group recommendations [45], we identified the subset of CD34+/VEGFR2+ EPCs.
Additionally, based on the literature data we assigned EPCs among the CD34+ fraction by the expression of Tie2, which is considered to be one of the major endothelial-specific receptor tyrosine kinases playing a central role in the formation of new blood vessels [46, 47]. Since CD34+/VEGFR2+ and CD34+/Tie2+ cells may also identify circulating mature endothelial cells shed from damaged vessels, subsequent works have included CD133 to distinguish EPCs from mature CECs [42, 45, 46].
In our study, we did not find significant differences in the values of CD34+/CD45(–)/CD133+ EPCs between patients with or without AECAs; however, AECAs positivity was associated with significantly lower values of CD34+/CD133+/VEGFR2+ EPCs as well as a significant decrease in those EPCs that express the Tie2 receptor (CD34+/CD133+/Tie-2+).
The population of CD34+/CD45(-)/CD133+ is called “true EPCs”, which are capable of forming highly proliferative late-outgrowth endothelial colonies (endothelial colony-forming cells – ECFCs) and behave as angioblasts with the capacity for endothelial differentiation and de novo vessel formation [21, 42]. They belong to a pool of vascular wall-resident rather than to a bone marrow-derived population [48]. In contrast, CD34+/CD133+/VEGFR2+ cells are designated as circulating angiogenic cells (CACs), are mostly bone marrow-derived and have potential to sustain angiogenesis but lack de novo vessel-forming activity. These cells better correlate with vascular endothelial status, have more potent vasoregenerative capacities and are functionally more potent with respect to homing and vascular repair [15, 21, 42, 44, 48, 49]. It was also demonstrated that EPCs expressing Tie2 show enhanced regeneration of denuded ECs monolayers compared to those without Tie2 expression, regardless of their VEGFR2 status [50]. Thus, the Tie2/angiopoietins signaling pathway in EPCs is essential to promote their angiogenic properties and induces mature endothelial phenotype of EPCs [46, 47, 50].
Combined with these data, our results may suggest that in SSc, AECAs target the bone-marrow derived “angiogenic” population of EPCs, thus affecting mostly angiogenic sprouting and endothelial repair instead of de novo vessel formation. Such possible AECA-associated decrease in Tie2+ and VEGFR2+ positive EPCs in SSc patients might be reflected in capillary collapse and progressive loss of angiogenesis, which may raise the possibility of more severe vascular manifestations in AECA-positive patients.
In fact, decreased circulating CD34+CD133+VEGFR2+ EPCs has been reported in SSc and correlated with severity of peripheral vascular manifestations, including impaired endothelium dependent vasodilation and active ischemic digital ulcers [16, 43]. The recent study by del Papa et al. may also support our suggestion. Authors have reported that SSc patients’ bone marrow plasma showed significant titers of AECAs, and their presence correlated with that of apoptotic EPCs [39]. These findings were further confirmed by an in vitro assay in which the apoptosis of normal progenitors was induced by the addition of AECA+ purified IgG [39].
It is likely that a portion of AECAs cross-react with EPCs as ECs develop from EPCs and may preserve some of their surface antigens (e.g. CD34). However, it is also possible that there is a novel population of anti-EPC antibodies distinct from ECs binding antibodies as it has been shown by competitive inhibition studies for patients with a differential risk profile for atherosclerotic vascular disease [38].
We realize several limitations of the study, including lack of male patients and healthy subjects as a control group, no clinical aspects of AECAs presence as well as in vitro assays of AECAs pathogenicity. Comparative analysis with other connective tissue diseases would also be appreciated. However, our results might be considered as preliminary data for further research in this field.
Conclusions
Our data provide evidence for an association between the presence of AECAs and the severity of endothelial damage in SSc as based on higher levels of total EMPs and CECs in AECA-positive subjects. In our study, AECAs were associated with ECs’ apoptosis and sloughing rather than their activation. We also pointed out the possible role of AECA positivity in impairment of vascular repair in SSc as evidenced by the significant decrease in the “angiogenic” population of EPCs expressing VEGFR2 or Tie2 receptors in peripheral blood of AECA-positive SSc patients.







