
Introduction
Behcet’s disease (BD) is a chronic, multisystem disease resulting from recurrent inflammation of the blood vessels (vasculitis). The usual symptoms include eye inflammation (uveitis) and recurrent aphthous-like ulcerations of the genital and oral mucosa. Other manifestations include neurological disorders, predominantly from the central nervous system (CNS), ulcerative skin lesions, arthritis and gastroenteritis [1, 2].
Although cases of BD can be found throughout the world, the vast majority occur in Turkey and countries in the Middle East and East Asia. In Europe, the disease is extremely rare in Caucasian patients. A recent Dutch study involving patients of different ethnic backgrounds showed a prevalence of 1/100,000 of the population among Dutch Caucasians, 71/100,000 among Turks and 39/100,000 among Moroccans [3]. Most studies have shown that BD affects males more often, with a peak in incidence in the second and third decades of life. In females, the severity of BD was shown to be less intensive [4, 5].
An association between the presence of the HLA-B-51 class I antigen and the pathogenesis of BD has been reported in the literature. The link between the B-51 and B-101 alleles and the occurrence of BD has been described in some Asian countries and in European patients with ocular symptoms. Although not a necessary factor for the disease, there is an association between HLA and the nature of the disease (Table 1) [1, 6].
Table 1
The aetiology of this disease is still unknown. It is presumed to be caused by some unidentified environmental factors, including bacteria, viruses or autoantigens that stimulate the immune system [1, 2]. The pathogenesis of BD is associated with increased production of pro-inflammatory cytokines, circulating immune complexes and autoantibodies directed against mucosal and endothelial cells [6–8].
Case report
A 56-year-old male patient was admitted to the Department of Oral Mucosal and Periodontal Diseases because of multiple ulcerative lesions located mainly in the oral cavity and on the facial skin. He reported general fatigue and malaise. The first symptoms appeared about 10 days prior to the visit, a few days after a dental procedure (extraction of multiple teeth). The patient linked the appearance of the first lesions directly to this procedure. As food and liquids were avoided due to pain, the patient’s general condition deteriorated.
The patient presents a general medical history of primary sclerosing cholangitis (PSC). In the course of several years of PSC, complications such as oesophageal mycosis and varices, secondary thrombocytopenia and ulcerative colitis appeared. Due to the progression of the disease, the patient is being prepared for liver transplantation. The patient denied other chronic diseases and had no family history and genetic disorders. Two weeks before the onset of the first symptoms, the patient was hospitalised for oesophageal varices in the Department of Internal Medicine and Haematology, where he was additionally diagnosed with oesophageal mycosis. During this hospitalization, laboratory tests were performed, the results of which were as follows: HIV antigen and antibodies – negative, HCV antibodies – negative, HBs – negative, tumour markers (AFP, CEA, CA 19-9, PSA) – negative. These tests were part of the diagnostic process in preparation of the patient for liver transplantation.
Upon examination, multiple painful ulcerations of approximately 1–2 cm in diameter were found to be present on the oral mucosa of the lips, cheeks, tongue, alveolar processes of the maxilla and the alveolar part of the mandible. The lesions were sharply demarcated, partially covered with fibrin and surrounded by an inflammatory border. The patient complained of pain in the entire oral cavity, submandibular area and throat. Deep ulcerative lesions with bloody or serous-purulent exudate, nodular and pustular lesions were also scattered on the trunk, limbs, facial skin (including the nasal cavity) and scrotum (Figures 1, 2). The lesions were very painful, with no pruritus, and the shallow ulcerations were covered by an eschar.
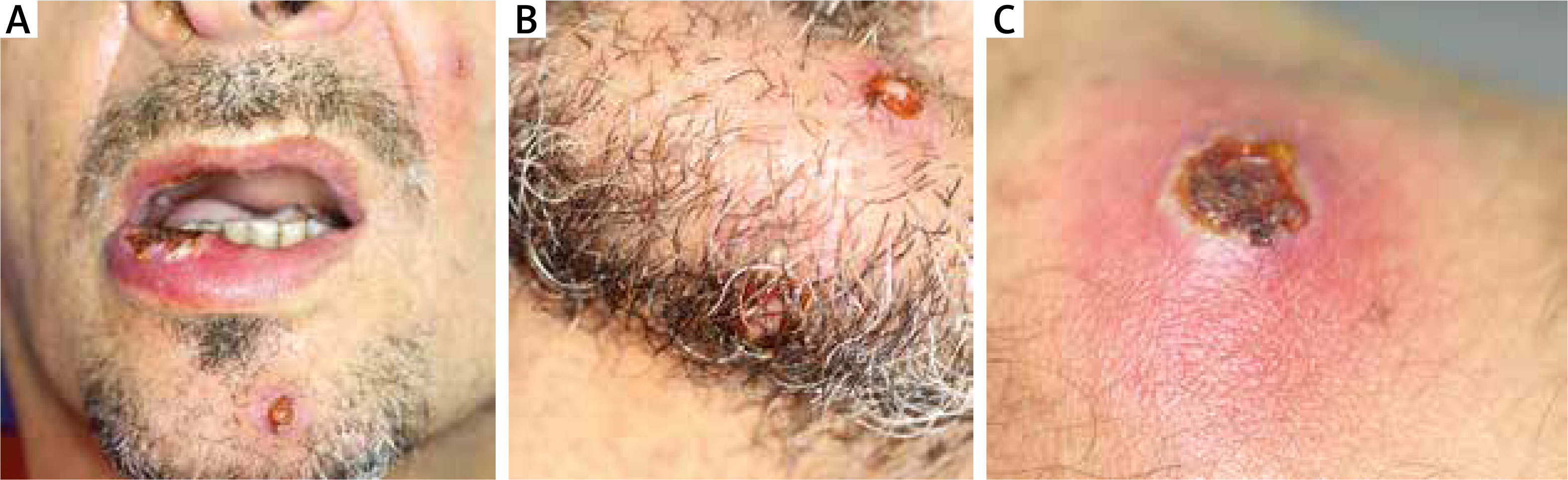
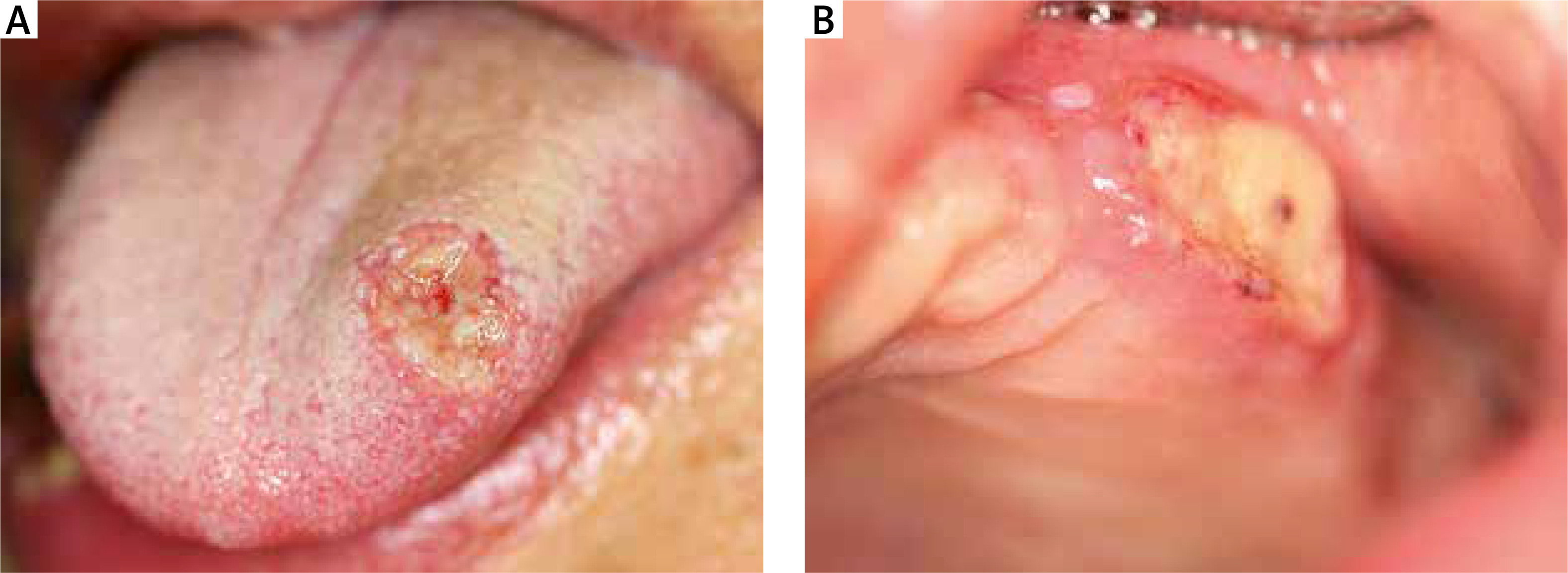
From the very beginning the skin and mucosal lesions were accompanied by symptoms of the upper respiratory tract infection and episodes of fever. Due to severe weakness and dissemination of lesions the patient was urgently referred to the Department of Dermatology, Medical University of Lodz.
Due to the ambiguous clinical picture, extensive diagnostics were performed during hospitalization in the Department of Dermatology. Microbiological examination of swabs from lesions on the skin and mucous membranes yielded no significant information. Staphylococcus epidermidis (probably due to contamination of the specimen with skin microbiota) and Streptococcus oralis, sensitive to vancomycin, were cultured. Blood cultures were sterile and the test for Treponema pallidum was negative. Biopsies were taken from the oral cavity and skin. A specimen taken from a lesion on the tongue showed an extensive and intense inflammatory infiltrate consisting mainly of neutrophils, a small number of macrophages and lymphocytes, and non-specific, indifferent granulation tissue. After 48 h, the pathergy test was positive. Ocular lesions were ruled out with an ophthalmological examination, and laryngological consultation also excluded any other disorder in the area of the head and neck.
As the clinical picture was suggestive of BD and the laboratory results excluded any other diagnosis, a definitive diagnosis of BD was made. The criteria were supported by the following symptoms: painful aphthous lesions in the oral cavity, ulcerations located in the genital area, abnormalities in the gastrointestinal tract with concomitant diarrhoea, and a positive test result for pseudofolliculitis (pseudofollicular inflammation).
The treatment consisted of antibiotic therapy, initially empirical (ceftriaxone i.v., 2 g/day) and then targeted (vancomycin, 2 g/day), as well as antifungal therapy (itraconazole, 100 mg/day) and immunosuppressive therapy (dapsone, 100 mg/day) in combination with dexamethasone 8 mg i.m. and then prednisone 40 mg p.o. Furthermore, treatment involved the intravenous fluid resuscitation, albumin replacement, potassium supplementation, torasemide and pantoprazole administration. Intensive topical treatment of skin lesions was also carried out, using skin spray with neomycin sulphate and dexamethasone, Pigmentum Castellani (active substances: phenol, resorcinol, boric acid), Formative Silver spray (colloidal silver and hyaluronic acid), mupirocin ointment, rinsing with saline solution. Lesions located in the oral cavity were treated with a gel containing flavones isolated from Scutellaria baicalensis root (Baikal thyroid), a diclofenac-based mouthwash (1 ml of solution contains 0.74 mg diclofenac), nystatin in suspension and a lotion with neomycin, hydrocortisone and anaesthesin. On the fourth day of hospitalisation, the patient’s condition improved, the fever subsided, and the lesions began to heal. Five weeks after the onset of the disease, the oral lesions were healed (Figure 3).
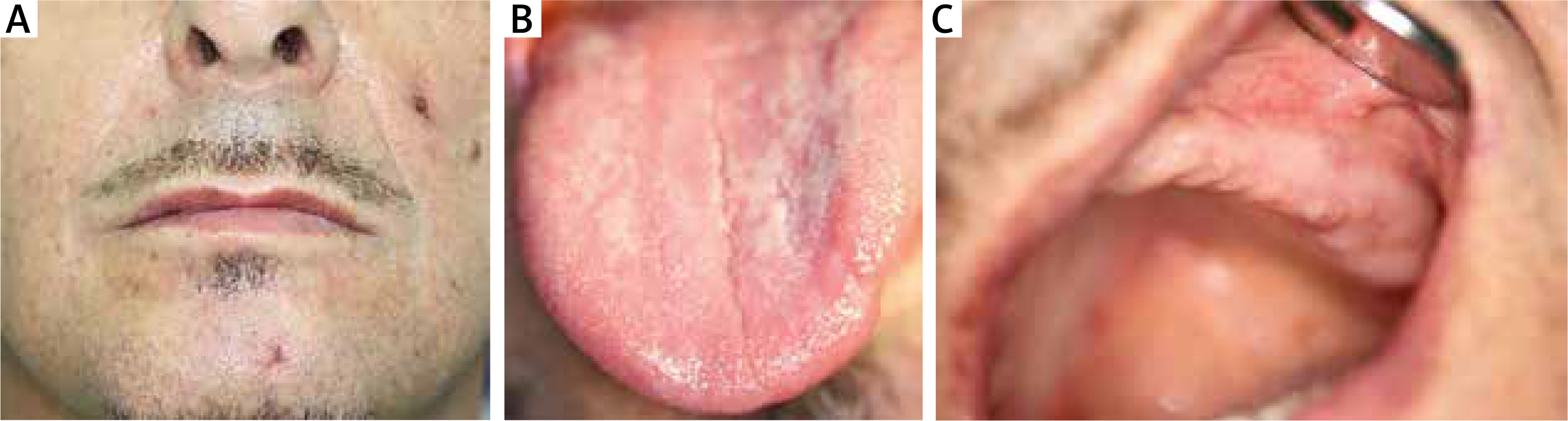
After hospitalisation, the patient continued treatment with cyclosporine (200 mg) and prednisone (20 mg). Two months after hospitalisation, the patient developed single ulcers, mainly located in the calf area. Modification of therapy at this stage consisted of higher doses of cyclosporine (300 mg) and prednisone (40 mg) together with topical application of Micro-Silver BG spray with highest purity micronized silver and ointment with anaesthesin. This therapy was continued for 3 months, after which it was replaced by cyclosporine (200 mg) and topical silver sulfathiazole cream. This treatment has continued to the present time and the patient reports only isolated ulcerations in the lower calf. The patient is currently being prepared for a liver transplant.
Discussion and literature review
BD occurs rarely in Central Europe; however, it is important for clinicians to be familiar with the symptoms, diagnostic methods and treatment. Typical symptoms include oral and genital ulcerations, skin lesions, inflammation of the eye area (uveitis, panuveitis, or retinal vasculitis) and many other systemic manifestations. Therefore, patients with BD require multidisciplinary care. The multitude of symptoms can contribute to diagnostic problems, so patients with initial symptoms may seek help from a dermatologist, dentist or ophthalmologist. Due to its severe course and numerous complications, this disease significantly affects the patient’s life. Sometimes, as a result of widespread multi-organ complications, it leads to disability and even death [4, 9].
Here we present a case of a patient with PSC who developed symptoms typical of BD. PSC is a chronic liver disease in which progressive fibrosis, destruction and narrowing of the intrahepatic and extrahepatic bile ducts result in cholestasis, which in turn leads to cirrhosis and liver failure. The aetiology of this disease is unknown, but autoimmune mechanisms are likely to underlie it. The peak incidence is between 30 and 50 years of age and the disease usually affects males, so the features are common to BD and PSC. Typically, patients report weakness, weight loss, excessive drowsiness and itchy skin, followed by jaundice. If inflammation of the bile ducts develops, fever may occur. Inflammatory bowel disease may coexist in most patients. Pharmacological treatment is limited due to the lack of knowledge about the cause of the disease. For severe abnormalities, an endoscopic procedure to dilate the bile ducts can be performed, but some patients require a liver transplant. It is likely (as with BD) that the disease may correlate with abnormal antigens of the HLA complex antigens, including those present on chromosome 6. The strong association with HLA suggests that HLA class I and/or class II genes are likely to be involved, and possibly HLA-B (class I) and DRB1 (class II) [10].
The presented case of a patient with PSC and BD may indicate a correlation in the pathogenesis of these diseases. The co-occurrence of both diseases may be genetically determined by the HLA system. Although different alleles are involved in the pathogenesis of the two diseases, the case described may suggest that further research is required. An additional argument for a potential link between these diseases is the hepatic manifestations observed in BD, such as sclerosing cholangitis and chronic hepatitis [10]. Alternatively, this patient developed an atypical form of BD, initially with sclerosing cholangitis as the only symptom. Several years later, after the diagnosis of PSC, a typical mucocutaneous form of BD occurred. To our knowledge, this is the first case in the literature to describe the diagnosis of BD in a patient with PSC.
In BD, the predominant clinical symptoms are mucocutaneous lesions. Aphthous-like ulcerations in the oral cavity are present in almost all cases. Oral lesions are the initial manifestation in up to 80% of patients and often precede later symptoms. In the oral cavity, it is characterised by numerous painful ulcerations, most often located on the non-keratinised oral mucosa – lips, buccal mucosa, ventral and lateral surfaces of the tongue, floor of the mouth and soft palate. Occurrences within keratinised mucosa such as the hard palate, gingiva and dorsum of the tongue are rare. Oral ulcers in BD are clinically indistinguishable from recurrent oral ulcers [11–15].
However, several features may be suggestive of BD, such as multiple ulcers (> 6), the presence of more than one clinical variant of ulcers at a time, involvement of the soft palate and oropharyngeal cavity. Genital ulcers are present in a significant number of cases, in males mainly localized on the scrotum. Occasionally, lesions may occur on the penis, in the urethra in men or in the vulva and vaginal area in women. Most patients present with skin symptoms [13–15]. The skin is usually covered with papulopustular or acneiform lesions. The patient in the present case had characteristic maculopapular skin lesions that showed a tendency to ulcerate. Disseminated lesions and multiple eruptions on the skin of the scrotum were also typical. Oral lesions were classified as minor aphthous ulcers. Some of the ulcerations were located in the most commonly affected areas, namely the lips, but some of the lesions found in this patient were located on the keratinised mucosa of the dorsum of the tongue and the alveolar process of the jaw [13–15].
Ocular lesions affect approximately 30–70% of patients and usually appear 2–3 years after oral and genital ulcers [9]. The lesions are usually severe and often bilateral. In the course of BD, uveitis, cataracts, glaucoma, vitreous inflammation, retinal detachment and oedema, cystic macular degeneration and closure of veins or arteries are observed [9]. A study by Kim et al. showed that the profile of intraocular chemokines differs in uveitis in patients with BD compared with other types of inflammation [16]. In the case described here, ocular involvement was excluded after ophthalmological consultation.
Neurological abnormalities in BD are mainly observed in males. Clinically, lesions may involve the brainstem and/or basal ganglia. Alternatively, non-parenchymal involvement includes cerebral venous thrombosis, arterial vasculitis, and aseptic meningitis [9]. Headache is the most common manifestation of BD and may be due to parenchymal or vascular changes, caused by increased intracranial pressure, meningitis or uveitis. Headaches unrelated to BD are also possible in patients with BD. Differentiating primary headache syndromes (migraine and tension-type) from neurological complications of BD can be difficult [17]. Magnetic resonance imaging is important to exclude multiple sclerosis. In our case, the patient had no neurological complications [9, 15].
Vascular disorders affect approximately 30–40% of patients with BD, especially males. Vascular complications such as superficial phlebitis, deep vein thrombosis, arterial thrombosis and aneurysms are most commonly seen. Cardiac complications are less common and include pericarditis, myocarditis, myocardial fibrosis, myocardiopathy and many others [13, 14].
Gastrointestinal symptoms in BD are occasionally described in the literature. The lesions may be localized in the whole gastrointestinal tract, but most frequently they occur in the ileocecal segment.
Abdominal pain, vomiting, diarrhoea, flatulence and gastrointestinal bleeding are symptoms reported by patients. Neutrophilic phlebitis and large vessel disease are the two most common forms of enteric disease in BD. Neutrophilic phlebitis leads to mucositis with ulcer formation. Later, intestinal ischaemia and intestinal infarction occur [13, 15, 16]. The most common manifestation of liver involvement in BD is Budd-Chiari syndrome (BCS), described as a blood flow disorder. It is a rare disease that is clinically manifested by a wide spectrum of symptoms, ranging from asymptomatic to fulminant liver failure. Mortality among patients with symptoms of liver disease remains high [13, 18]. Oesophageal involvement is rare and manifests as ulcerations, mainly in the middle or distal part of the oesophagus. Typical symptoms include retrosternal pain, dysphagia, odynophagia and melena. Complications of oesophageal involvement include strictures and perforations. In addition, indigestion, epigastric pain, gastric or duodenal ulcers may occur in the course of BD. In our patient, exacerbation of intestinal disease and deterioration of liver function parameters were observed.
Differential diagnosis
Due to the lack of typical laboratory diagnostic markers, the diagnosis of BD is mainly based on clinical symptoms. To diagnose BD, several criteria must be met [4]. The criteria of the International Study Group for Behcet’s Disease (ISG) 1990 or the International Criteria for Behcet’s Disease (ICBD) 2013 (Table 2) can be used [12].
Table 2
Diagnostic criteria for Behcet’s disease. The International Criteria for Behcet’s Disease (ICBD) were updated in 2013, as a replacement for the International Study Group for Behçet’s disease (ISG) [12]
| ISG | ICBD |
|---|---|
|
Oral ulcerations in BD can be clinically similar to herpetic lesions or zoster lesions, recurrent aphthous ulceration (RAU), erythema multiforme (EM) and many other diseases. Herpes simplex ulcers are clustered, small and shallow. They are usually recurrent and appear in the same location, mainly on the keratinised oral mucosa. The Tzanck test (microscopic examination of smears of cellular material taken from hair follicles) may be helpful in the differential diagnosis [12, 19]. RAU and classical EM are characterised by the absence of systemic symptoms. In EM, target-like lesions appear on the skin. In RAU, lesions are found only in the oral cavity [12].
Lesions associated with tuberculosis should also be considered in the differential diagnosis. Chest radiography can be helpful in the diagnostic process, with lung involvement found in 32% of patients with intestinal tuberculosis (ITB) [13].
Inflammatory bowel diseases, mainly Crohn’s disease (CD), should also be considered in the differential diagnosis of BD. Endoscopic biopsy may be helpful. Differentiation between BD and CD is quite difficult due to the similar time of onset (young people) and similar symptoms, including gastrointestinal and extraintestinal abnormalities [5, 13, 20].
Rare immune-mediated syndromes, characterised by increased production of inflammatory cytokines and lack of detectable autoantibodies, should also be considered in the differential diagnosis.
Mevalonate kinase deficiency (MKD) is an autosomal recessively inherited disease in which there is insufficient activity of mevalonate kinase (involved in cholesterol biosynthesis). Symptoms include recurrent fever, abdominal pain, diarrhoea, nausea, arthritis, joint pain, headaches and cellulitis. MKD can manifest as a maculopapular rash and oral aphthae which occur in half of patients [21].
In Sweet’s syndrome there are skin lesions similar to BD. Not only tender erythematous nodules or plaques are observed, but also ulcerations and blisters. Oral lesions are very rare. In addition to skin and mucosal manifestations, fever and neutrophilia are observed [5, 15, 22–24].
Blood cell counts and biochemical tests are useful in determining the severity of BD. Magnetic resonance imaging (MRI) may be helpful in detecting abnormalities in cases of CNS involvement [25].
Systemic treatment
Management of BD is difficult due to the plethora of symptoms involving multiple systems. Although there are some suggested treatment regimens, there are no clear protocols covering the treatment of vascular, neurological and gastrointestinal symptoms.
Immunosuppressive therapy and corticosteroids (CS) are commonly used in the treatment of BD. CS are mostly used in moderate to severe cases and the dose depends on the severity of symptoms. Doses of 20 –100 mg prednisolone or weight-dependent doses of 0.5–1 mg/kg per day for 1–2 weeks are used, then reduced by 5 mg per week until treatment is discontinued. Another steroid used to treat BD is methylprednisolone at 1 g/day for 3 days, followed by prednisone at 1 mg/kg/day at slow weaning [26]. It is important to remember that complete remission is achieved with CS therapy in about half of cases, and some patients do not respond well to it. Maintenance treatment with corticosteroids is not recommended and long-term use should be avoided due to significant systemic side effects.
Usually, CS therapy should be supplemented with immunomodulators or immunosuppressive drugs.
CS are often combined with azathioprine and/or colchicine [1, 8, 13]. Monotherapy with immunosuppressive drugs is the treatment of choice for patients with steroid-dependent or steroid-resistant intestinal BD [13]. Thalidomide, mycophenolate, cyclosporine, methotrexate, interferon tacrolimus and TNF-α antagonists have also been used in the treatment of BD. The clinical efficacy of azithromycin, isotretinoin and acyclovir has also been described (Tables 3, 4) [13, 24].
Table 4
Colchicine is commonly used in patients with mucocutaneous symptoms and arthritis in BD [12]. In the treatment of oral ulcers, 0.6–1 mg can be administered 2–3 times a day. The mechanism of action of colchicine is to inhibit neutrophil chemotaxis. However, its efficacy is limited [23, 25].
Dapsone has a complex mechanism of action that includes inhibiting neutrophil chemotaxis, similar to colchicine. In clinical trials, dapsone treatment was shown to result in less frequent occurrence and faster healing of oral ulcers, as well as a reduction in skin manifestations. Dapsone has also been shown to reduce pathergy reaction. Dapsone is usually used at a dose of 100 mg/day [13, 25].
Cyclosporine A can be also used in the patients with mucocutaneous manifestations. However, cyclosporine A therapy has potential limitations and contraindications, and side effects are common [9, 13, 23, 24].
Thalidomide administered at a dose of 100–300 mg/day has been used to treat orogenital ulcers. Although the efficacy of thalidomide is relatively high and has been shown to completely resolve symptoms within a few weeks, it can cause potential side effects including paraesthesia, drowsiness, nausea, and vomiting [12, 13, 15, 22–25].
Etanercept, a TNF-α inhibitor, has been shown to be of benefit in BD. The therapy reduced mucocutaneous symptoms, including oral ulcers, but without suppressing the pathergy reaction. The recombinant human monoclonal antibody adalimumab had similar effects [9, 12, 13, 23, 25].
Topical treatment
Topical agents with lidocaine, chlorhexidine, triclosan or silver nitrate are used for symptomatic treatment of RAU [9]. Patients should be given recommendations to improve oral hygiene and avoid acidic, salty and spicy foods [9, 24].
CS are widely used for topical therapy of oral and genital lesions [26, 27]. The most commonly used CS for the treatment of RAS is triamcinolone acetonide 0.1% in a twice-daily paste. Topical dexamethasone elixir (0.5 mg/5 ml) may be helpful in controlling hard to reach or multiple ulcers. Severe lesions require stronger steroids: betamethasone sodium phosphate rinse (0.5 mg/5 ml), beclomethasone dipropionate spray (100 μg/puff), clobetasol orabase 0.05% or fluocinonide orabase 0.05%. In the most severe cases, intradermal injection of CS may be considered. The advantage of this method is to obtain a high concentration of the drug at the lesion site with minimal systemic absorption. Triamcinolone (5–10 mg/ml, administered at 0.1–0.5 ml per lesion) can be used [15, 28].
A topical suspension of sucralfate (5 ml 4 times daily) may be used in the treatment of gastrointestinal and oral ulcers. Sucralfate stimulates skin fibroblast proliferation and formation of the granulation tissue, contributing to improved healing and formation of a specific protective barrier.
Clinically, a reduction in pain and healing time was demonstrated. Some authors suggest that sucralfate suspensions can be used as prophylaxis against the development of oral ulcers in patients with BD. This substance can also be used to treat lesions in the genital area [9, 12, 15].
In the treatment of skin lesions, improvement is obtained by using pimecrolimus cream. It is a selective inhibitor of calcineurin synthesis and secretion with a strong anti-inflammatory effect [9, 24].
Conclusions
BD is a rare systemic inflammatory disease of unclear aetiology. A history of recurrent, severe oral and skin ulcers may be helpful in the diagnostic process. As oral ulcerations sometimes precede other symptoms of BD, the disease may remain undiagnosed for months/years in some patients. The clinical examination should be complemented by a comprehensive list of laboratory tests to help rule out other causes of symptoms. Comorbidities further add to the complexity of the diagnostic process. In this case, BD was diagnosed in a patient with PSC. There is no evidence of other cases of co-occurrence of these conditions. The treatment of BD includes systemic immunosuppressive drugs and topical antimicrobial and antibacterial agents.







