
Introduction
In the recent years, a great number of reports concerning the keratins 5 and 14 gene mutations in epidermolysis bullosa simplex, and keratin 1 and 10 gene mutations in ichthyoses have been published, making great strides in understanding both the basic biology of human keratins and the aetiology and pathogenesis of a number of specific human single gene disorders. Keratins are products of a large family of genes encoding intermediate filaments. The protein products of these genes constitute the largest component of keratinocytes, about 85% of cellular proteins. Keratin binds into intermediate filaments without the participation of cofactors other than additional keratin – they polymerize only as heterodimers and always consist of equimolar amounts of acidic (type 1) and alkaline (type 2) keratins. In human epidermis, keratins type 5 and 14 are expressed in the basal layer, whereas keratins 1 and 10 in the stratum spinosum.
Mutations in the gene encoding keratin 1 (KRT1) cause epidermolytic hyperkeratosis – a rare autosomal dominant pathology of cornification. It was originally termed bullous congenital ichthyosiform erythroderma owing to the hallmark features of erythroderma, blistering and skin denudation present at birth and subsequent development of marked hyperkeratosis. This presentation occurs with palmoplantar keratoderma. Epidermolytic hyperkeratosis is easily distinguishable from the other forms of congenital ichthyoses via its highly characteristic histologic findings, which include dense hyperkeratosis, hypergranulosis, coarse keratohyaline granules and cytolysis leading to the suprabasal blister formation. Keratinocytes demonstrate marked intracellular vacuolar degeneration and dense clumps of keratin intermediate filaments. Varying degrees of dyskeratosis and a mild perivascular lymphohistiocytic infiltrate may also occur [1].
Epidermolytic hyperkeratosis usually presents soon after birth. Treatment is primarily symptomatic and depends on the patient’s age and presentation. Infants should be monitored in the intensive care setting to manage dehydration, electrolyte imbalance, and cutaneous superinfection.
Delays in diagnosis and management can lead to negative outcomes and possible death [1].
Skin fragility manifests with diminished resistance of the skin to external mechanical forces and with skin blistering, erosions, and painful wounds as clinical features. Skin fragility disorders are caused by mutations in several distinct genes that encode proteins involved in epidermal integrity and dermal–epidermal adhesion. The genetic spectrum, along with environmental and genetic modifiers, creates a large number of clinical phenotypes, spanning from minor localized lesions to severe generalized blistering or early demise resulting from extensive loss of the epidermis, these cases often turned out to be a mutation in KRT1 [2]. Another skin condition caused by genetic changes in the KRT1 gene, called cyclic ichthyosis with epidermolytic hyperkeratosis, is similar to epidermolytic hyperkeratosis, but the skin changes disappear for short periods, then recur. The recurrent skin changes can last for weeks or months [3].
Epidermolysis bullosa simplex (EBS) is characterized by superficial, intraepidermal blister healing without scars. In the common dominant EBS subtypes caused by mutations in the genes encoding either keratin 5 or keratin 14, blisters develop within the basal keratinocyte layer, whereas there is a growing body of evidence that in the rare EBS variants, blisters develop suprabasally. Pathogenic mutations in more than 20 different genes have been identified as a molecular cause of EB [4–6]. However, the list is certainly not closed and novel reports indicating contribution of other genes to EB, especially to suprabasal EBS etiopathology, emerge [7].
Herein, we present four patients with an epidermal barrier defect manifesting by blistering due to the KRT1 mutations: two with a severe form of epidermolytic hyperkeratosis at the different stage of the disease and the case of suprabasal EBS, which, to the best of our knowledge, is the first description of EBS caused by a mutation within the KRT1 gene.
Aim
The aim of the study was to analyse the phenotypic spectrum of blistering disorders caused by the KRT1 mutations. We present the extensive characteristics of histopathological, immunofluorescence and electron microscopic features together with assessment of possible differences between these clinically distinct disorders.
Material and methods
Four patients with epidermal barrier defect manifesting as blistering with the KRT1 mutations were included to the study. Final clinical diagnosis was established according to clinical, histopathological immunofluorescence and electron microscopic evaluation.
Results
Case 1
The patient, the first and only child of healthy parents, was born with two erosions on the abdominal region and thigh (Figure 1 A) rapidly spreading all over the entire skin with bullae, including some well tensed and positive Nikolsky sign. Within month after birth, cerebral haemorrhage of the second degree/class II cerebral haemorrhage and seizures have developed in association with sepsis. The degree III retinopathy was diagnosed at the age of 1 month. Pronounced palmoplantar hyperkeratosis started at the age of 6 months eventually leading to hand contractures.
Figure 1
A – Girl at the age of 3 months with superficial erosion on the leg. B – Patient at the age of 16 with large erosion on the back. C – Histology shows dense hyperkeratosis, hypergranulosis, coarse keratohyaline granules and intracellular vacuolar degeneration and varying degrees of dyskeratosis and a mild perivascular lymphohistiocytic infiltrate. D – Electron microscopy shows dense clumps of keratin intermediate filaments keratin with in stratum spinosum
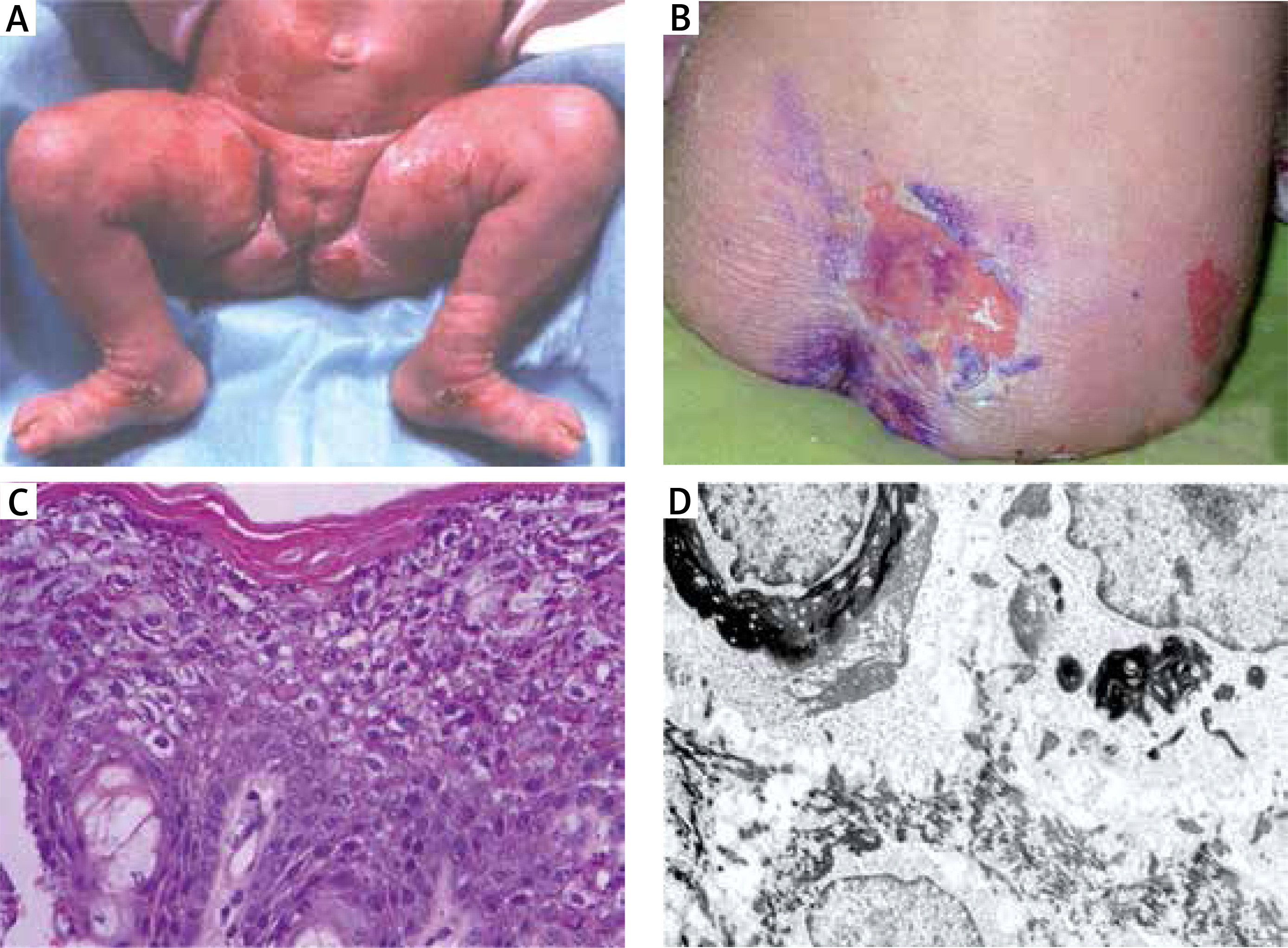
At the age of 16 she was seen with psoriasiform erythroderma alternating with atopiform dermatitis erythroderma with scaling. Large, flaccid, purulent blisters and erosions with no predilection to joints areas were noticed (Figure 1 B). The hair were normal. Unspecific nail changes with mild subungual hyperkeratosis and Beau’s lines were seen. Mucous membranes were unaffected and ectropion was not present.
Histology showed pronounced hyperkeratosis and disruption of cytoplasm in keratinocytes of the stratum spinosum and keratin clumping (Figure 1 C).
An electron microscopic study of perilesional skin confirmed clumping of keratin intermediate filaments and cytolysis of keratinocytes of the stratum spinosum (Figure 1 D).
Based on the clinical picture and microscopic analysis, the patients were diagnosed with epidermolytic hyperkeratosis and the molecular analysis revealed the mutation p.Asn188Lys within exon 1 of the KRT1 gene leading to a change from asparagine to lysine.
Case 2
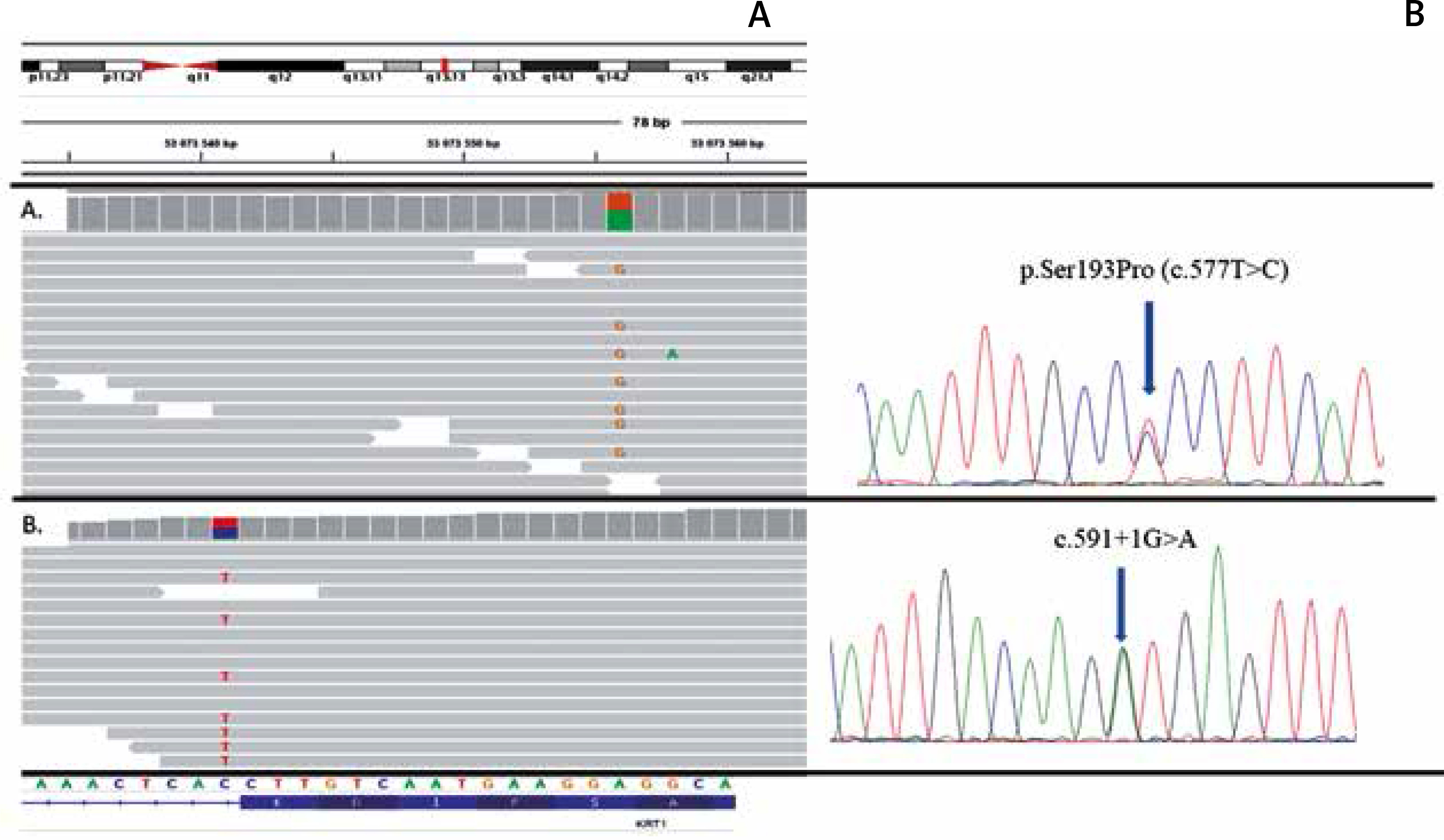
A child of healthy parents was born with widespread blisters and erosions, clinically diagnosed as generalized epidermolysis bullosa. The girl died 4 days after birth due to sepsis as a result of the disruption of the epidermal barrier. Molecular analysis using next-generation sequencing of epidermolysis bullosa causative genes panel did not reveal any mutation in the coding regions of the following genes: COL17A1, COL7A1, DSP, DST, EXPH5, FERMT1, ITGA6, ITGB4, JUP, KRT5, KRT14, LAMA3, LAMB3, LAMC2, PKP1, PLEC1 and TGM5. However, the extended NGS analysis disclosed de novo pathogenic mutation p.Ser193Pro within the KRT1 gene in the proband (Figure 2).
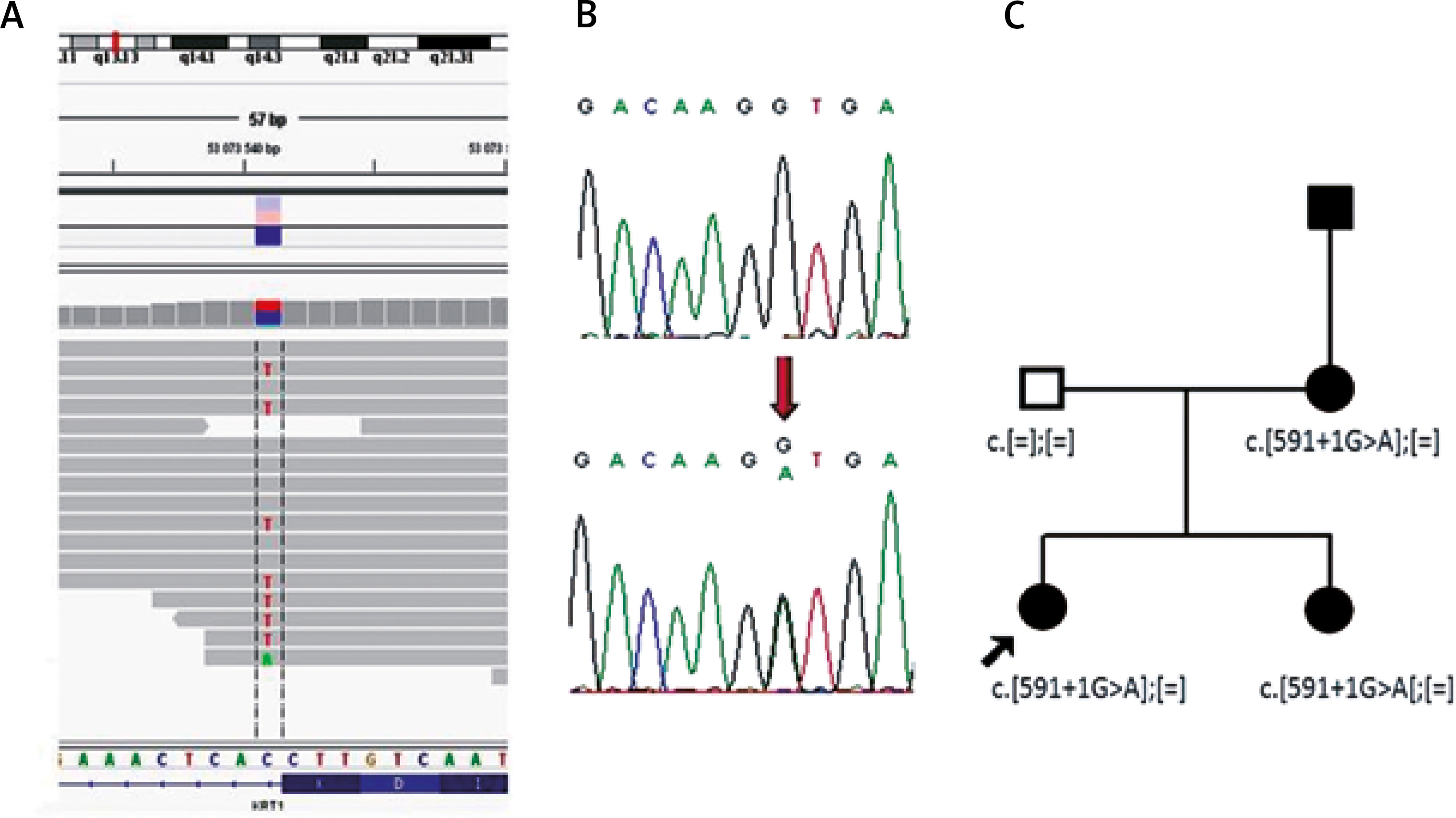
Figure 2
Results of molecular analysis revealing mutations p.Ser193Pro and c.591+1G>A. Top- chromosomal localisation of the KRT1 gene; bottom sequence of the KRT1 gene with exon-intron border. A – Results of p.Ser193Pro detection using NGS and Sanger sequencing. B – Results of c.591+1G>A detection using NGS and Sanger sequencing
Cases 3 and 4
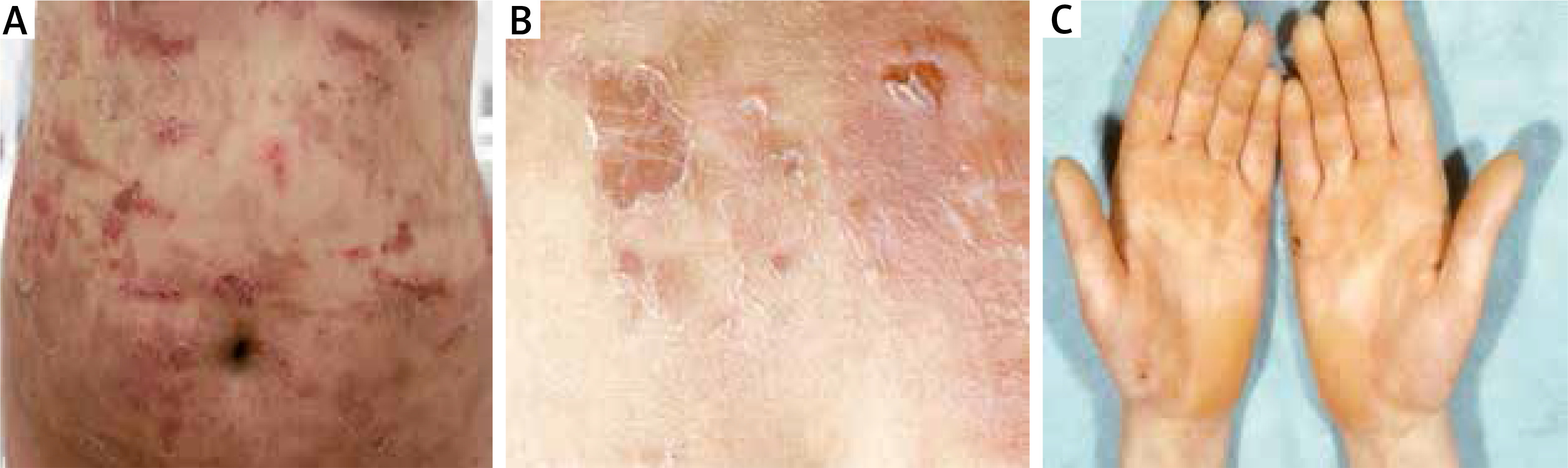
A twenty-four-year-old woman and her 1-year-old older sister initially presented to our department in 2005 with generalized flaccid blisters. In both sisters, the blisters and erosions were localized in the extremities and trunk with predominantly involved lower abdomen due to mechanical friction with clothes (Figure 3 A). Erosions healed without scars and milia, but some of them left post-inflammatory hyperpigmentation. Nikolsky sign was positive on the trunk and lower abdomen (Figure 3 B). Palmoplantar hyperkeratosis was observed in both patients without blisters on hands and feet (Figure 3 C). Family history revealed that during a 12-year follow-up both sisters presented severe relapses of the disease several times a year with short periods of remission. Bullous lesions developed in both sisters at birth and the course of the disease was particularly severe in the neonatal period. Moreover, the patients’ mother and grandfather manifested blisters at birth and during childhood, whereas palmoplantar hyperkeratosis persisted in adulthood.
Figure 3
A – Multiple trauma-induced erosions on the abdomen healing without scars but leaving hyperpigmentation, B – superficial blisters and Nikolsky sign on the lower abdomen, C – keratoderma on the hands
Histology showed orthokeratosis and disruption of cytoplasm in keratinocytes of the stratum spinosum leading to vesicle formation and intraepidermal separation (Figure 4 A).
Figure 4
A – Histology shows orthokeratosis and disruption of cytoplasm in keratinocytes of stratum spinosum leading to vesicles formation and intraepidermal separation. B – Double immunofluorescence mapping shows intraepidermal separation through stratum spinosum of the epidermis leaving normal basal keratinocytes attached to the dermal-epidermal junction labelled with both basal membrane zone markers
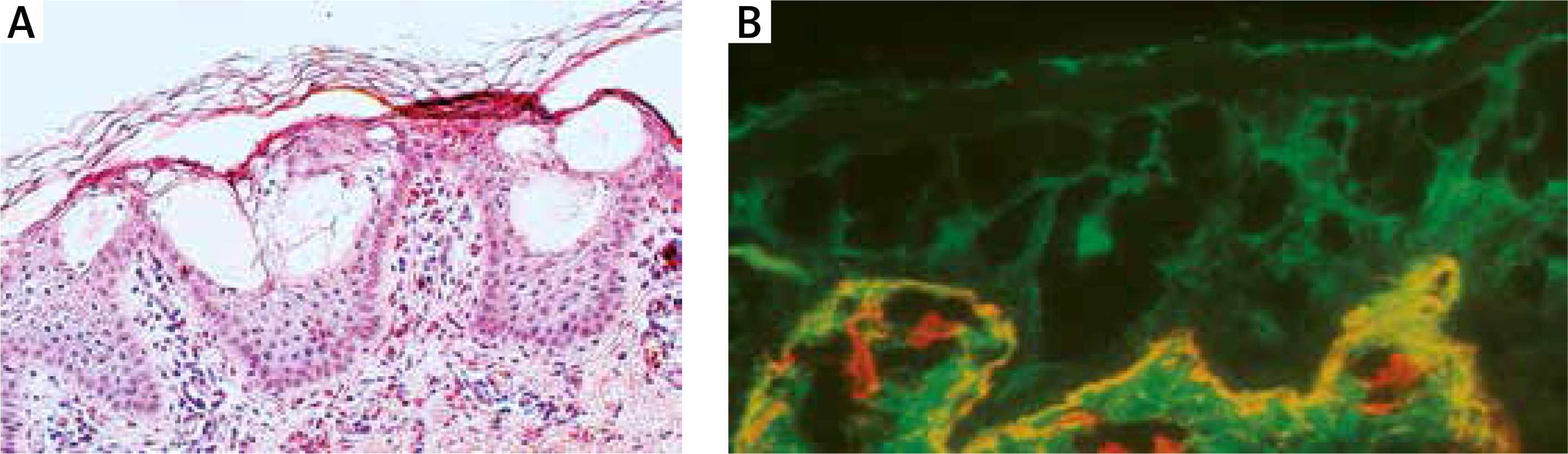
Double immunofluorescence mapping was performed on biopsy samples of apparently normal skin close to the fresh blister and perilesional skin, using antibodies directed to the lamina densa (monoclonal antibody against collagen IV Sigma, clone 7.2) and the upper part of the lamina lucida (bullous pemphigoid serum against BP180 NC16A – positive, and BP230 – negative). Intraepidermal separation through the stratum spinosum of the epidermis was observed leaving normal basal keratinocytes attached to the dermal-epidermal junction labelled with both basal membrane zone (BMZ) markers (Figure 4 B). An electron microscopic study of perilesional skin showed clumping of keratin intermediate filaments and cytolysis of keratinocytes of the stratum spinosum (Figure 5). Based on the clinical picture and microscopic analysis, the patients were diagnosed with EBS, but the molecular analysis of KRT5 and KRT14 did not show any mutations (Sanger Sequencing, according to Hamada et al.) [2]. Recently, next-generation sequencing (NGS) was performed (using TruSight One Panel, Illumina) showing the heterozygous c.591+1G>A mutation within the KRT1 gene encoding keratin 1, leading to a change from guanine to adenine in a highly conserved donor splice site position of intron 1.
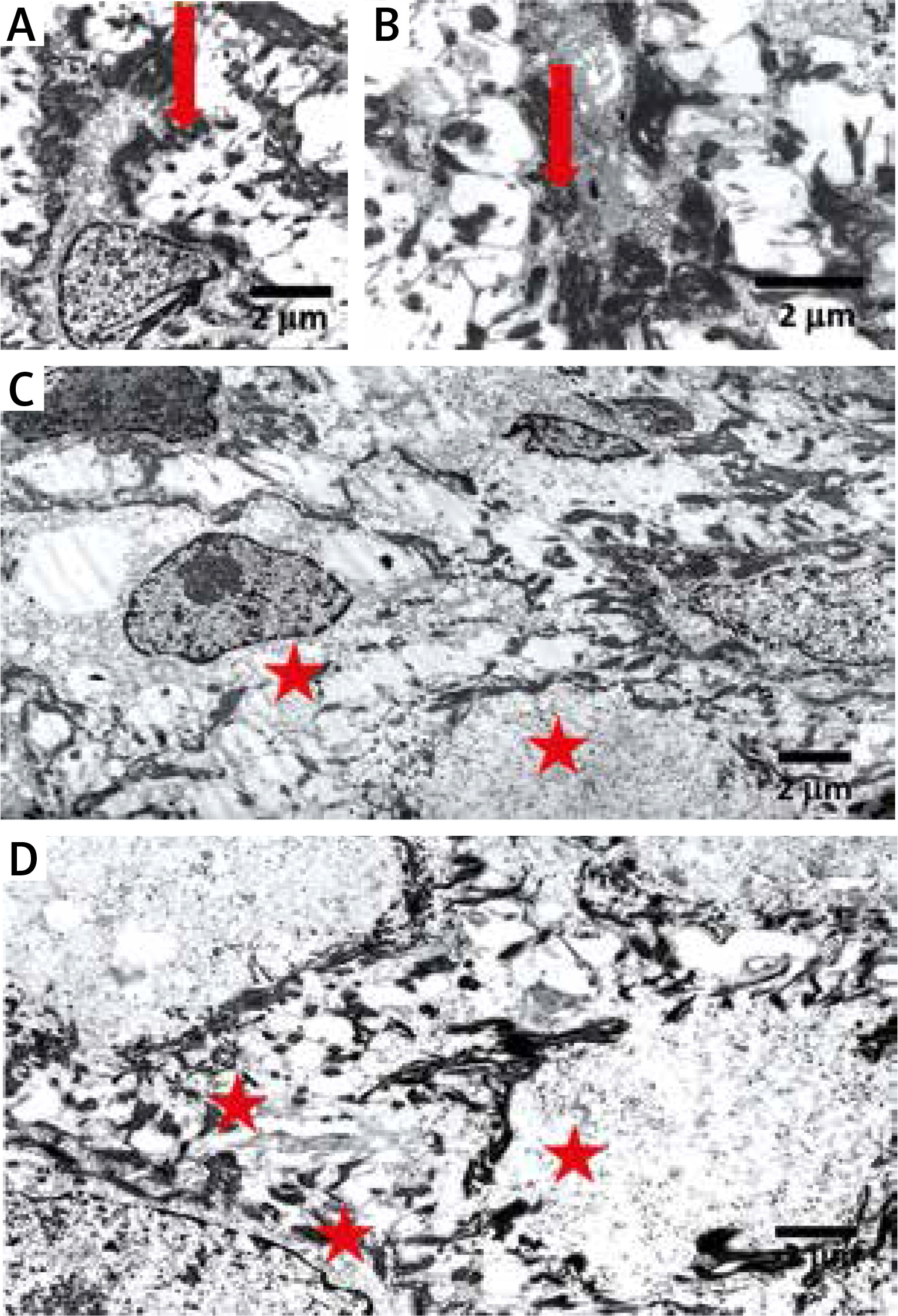
Figure 5
Electron microscopy shows clumping of keratin intermediate filaments (arrows in A, B) and cytolysis in the keratinocytes of stratum spinosum (asterisks in C, D). Bars: 2 μm
The presence of c.591+1G>A mutation in both sisters and their mother was further confirmed by Sanger sequencing (Figure 6). The DNA of the affected grandfather was unavailable.
Discussion
We studied four patients with KRT1 pathology who present blisters. Patient 1 represents epidermolytic hyperkeratosis – bullous ichthyosiform erythroderma, genetic disorder characterized by blistering in the neonatal period followed by ichthyotic hyperkeratosis in childhood and adolescent life. Hyperkeratosis in adolescence that replaces the blisters and erosions is the result of compensation of the epidermal barrier defect. Interestingly, this patient develops blisters also in adolescence, which reflects the severe course of the disease. Molecular analysis, histology and electron microscopic findings are consistent with the diagnosis of epidermolytic hyperkeratosis.
A newborn child with extensive blisters and erosions who died 4 days after birth was clinically misdiagnosed as lethal generalized epidermolysis bullosa and the proper diagnosis of epidermolytic hyperkeratosis is based on molecular analysis. The child had p.Ser193Pro mutation in one allele of the KRT1 gene, which occurred de novo, as shown by a comparative molecular test with DNA of the child’s parents. The diagnosis was made post mortem and the conclusive result was only possible by the implementation of next-generation sequencing using a panel of both EB and other fragile skin disorder-related genes. The KRT1 mutations are known from being causative for a rare epidermolytic hyperkeratosis, which may be fatal in newborns. However, in contrast to lethal generalized epidermolysis bullosa, if only the child survived this period, the prognosis is rather good [8]. Hence, by presenting this case, we would like not only to show another face of KRT1 molecular defects, but also to illustrate the importance of profound diagnostics. In this case, the risk of having another affected child for the parents was diminished from 25%, as calculated initially for EB, to very low.
The most interesting part of our study was to determine the mutation which triggered bullous genodermatosis in our 36-year-old patient and her family. Clinical observations and molecular studies were conducted for the first time in 2005. Immunofluorescence mapping analysis revealed intraepidermal blister formation characteristic of EBS, ruling out dystrophic and junctional EB. The mode of inheritance of the disease and the presence of palmoplantar hyperkeratosis along with flaccid blisters on the trunk and extremities strongly suggest keratin pathology. Nonetheless, molecular analysis of the KRT5 and KRT14 genes did not show any mutations. Almost 10 years after the first molecular efforts it was possible to identify the KRT1 heterozygous c.591+1G>A mutation using a novel and powerful molecular strategy – next-generation sequencing – allowing for a rapid analysis of several genes. This modern diagnostic method seems to be extremely useful in the molecular analysis of genodermatoses in which several candidate genes are linked to similar pathology, e.g. different variants of ichthyosis or EB [9]. More importantly, in contrast to previously performed diagnostic procedures, more genes can be easily co-analysed, enabling the identification of mutations in genes other than those selected initially by phenotype evaluation. Consequently, novel, yet unexpected, phenotype-genotype observations can be made. To the best of our knowledge, it is the first report of a family with EBS caused by the KRT1 mutation. Of note, the level of blister formation in the stratum spinosum reflects keratin 1 pathology. It is well known that KRT1 mutations usually cause a bullous variant of ichthyosis – a genodermatosis in which blisters appear at birth and in the neonatal period, but in childhood the skin lesions transform into ichthyotic scales and palmoplantar hyperkeratosis [10]. In contrast to the typical picture of bullous ichthyosis, none of the family members manifested scaly skin. However, Nikolsky sign is present and flaccid blisters develop continuously after a mild trauma and persist in adulthood. Moreover, histopathology showing blister formation through the stratum spinosum, suggestive of suprabasal EBS, and no signs of hyperkeratosis ruled out bullous ichthyosis. It is well known that depending on the type of mutation and its localization within the gene, the clinical picture of certain genodermatoses may be strikingly different or even represent a distinct entity. For instance, a mutation in the tail of the KRT1 gene causes ichthyosis with confetti clinically characterized by scaling erythroderma and white spots, due to revertant mosaicism; however, blisters never occur in this disorder, contrary to bullous ichthyosis [11]. On the other hand, KRT5 and KRT14 mutations are strongly associated with EBS, whereas the loss of function mutations within KRT5 are mostly related to reticulate pigmentation without blister formation (Dowling-Degos disease) [12]. Finally, gene mutations in different types of epidermal keratins cause epidermolysis bullosa simplex, epidermolytic ichthyosis, superficial epidermolytic ichthyosis, epidermolytic palmoplantar keratoderma and pachyonychia congenita/focal palmoplantar keratoderma, which present with thickening of the palms and soles with underlying blister formation. The particular mechanism of blister formation in keratinizing diseases has not been well established, but we suggest that the compensation of an epidermal barrier defect may be the most reasonable explanation [13]. Our patients present a novel hereditary blistering disorder due to keratin 1 pathology confirmed by the disruption of cytoplasm in the stratum spinosum in histology, and clumping of keratin filaments at the ultrastructural level. Although this is the first study which associates KRT1 with EBS pathology, it is necessary to stress that the heterozygous c.591+1G>A mutation in the KRT1 gene was previously reported in one patient diagnosed with a bullous variant of ichthyosis [14]. The clinical details of a Swedish patient were not presented by the authors, who focused their study on a molecular analysis of 15 ichthyotic cases. It remains unclear why the same mutation causes two different entities – bullous ichthyosis in the Swedish patient and EBS in our Polish family. One explanation may be that the Swedish patient clinically presented palmoplantar hyperkeratosis without blisters in adult life just as the mother of our two EBS sisters. It is also possible that some genetic or environmental factors may influence the course of genodermatoses.
In contrast to the Swedish authors, we present a 13-year follow-up of two adult sisters constantly developing blisters induced by mechanical trauma, without ultrastructural and histological signs of ichthyosis. Therefore, we propose including the heterozygous c.591+1G>A mutation in the KRT1 gene in the list of molecular defects underlying EBS pathology.
According to HGMD, there are 70 distinct mutations in KRT1 genes, which includes 46 missenses and 6 splicing mutations. There were several attempts to establish strict genotype-phenotype correlations among the genes encoding keratins and clinical outcome of their mutations. As stated above, the basic structural unit of intermediate filaments are dimers, which are made of two interacting keratins. Therefore, one of the mostly discussed factors influencing the severity of the mutation is the position of amino acid in the region where two keratins interact on a molecular level. Indeed both missense mutations: p.Ser193Pro and p.Asn188Lys are localized within highly conserved helix initiation motif (HIM). Furthermore, the p.Ser193Pro was reported before to cause severe PPK and generalized hyperkeratosis in another family where it also occurred de novo [15]. The mutations of codon 188 leading to different amino acid substitutions are recurrent. According to the Intermediate Filament Database, two other patients with p.Asn188Lys substitution were described in the literature, the one with diffuse thick keratoderma on both palms and soles with moderate scaly patches on the entire body and another one with PPK and blisters [16, 17].







