
Interstitial granulomatous dermatitis (IGD) is a rare dermatological entity belonging to a group of non-infectious cutaneous granulomas and is considered to be one of subtypes of the cutaneous reaction pattern known as reactive granulomatous dermatitis (RGD). Other subtypes of this condition are palisaded and neutrophilic granulomatous dermatitis (PGND) and interstitial granulomatous drug reaction (IGDR). Although there are differences in the literature, mainly in the histopathological picture, described between various subtypes of RGD, this division is becoming less unambiguous with each newly described case due to overlapping spectra of clinical symptoms. IGD in many cases is associated with various autoimmune diseases, primarily with rheumatoid arthritis [1]. There were also reports of its association with bacterial, parasitic, neoplastic diseases as well as drug-induced cases were described [1, 2]. We present 2 cases of IGD of atypical clinical picture that could suggest initial diagnosis of PGND as well as an updated literature review of the clinical presentation, histopathological features and management of this rare entity.
One of described patients (case 1), a 72-year-old female, came to our Dermatology Department with a three-year history of asymptomatic erythematous lesions with violaceous hue, overlying, accompanied with deep seated, hard skin nodules (Figures 1 A, B). Additionally, there were small, hard, non-painful, movable nodules of approx. 5 mm in diameter palpable within lesions on knees and elbows. There were no comorbidities and no medications were taken. Two skin biopsies were taken for histopathological verification of the diagnosis.
Figure 1
Clinical and histopathological presentation of case 1. A, B – asymptomatic erythematous lesions with violaceous hue, overlying joints, and accompanied by deep seated, hard skin nodules. C – interstitial lymphohistiocytic infiltrate with marked neutrophile presence (H + E, 400×), D: dispersed mixed inflammatory infiltrate in the upper dermis changing into interstitial granulomatous infiltrate in the mid-dermis (H + E, 100×)
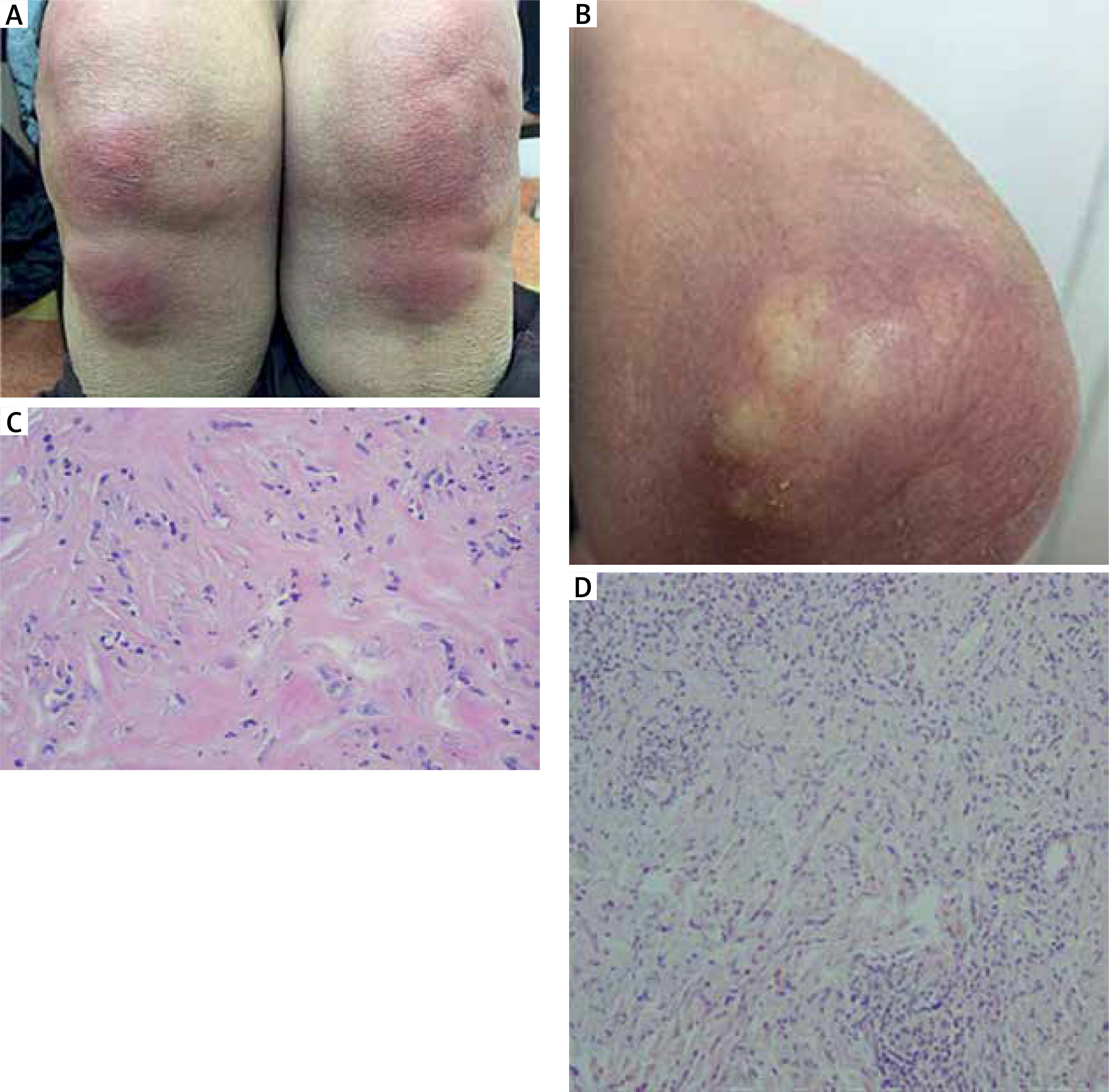
In examination of the first one, taken from the skin lesion overlying metacarpophalangeal joint of the right hand, revealed signs of lichenoid dermatitis with a thin epidermis covered with thick, ortokeratotic stratum corneum with multiple cytoid bodies and vacuolization of the basal layer. Under the epidermis, an abundant, dispersed inflammatory cell infiltrate was observed and so were dilated blood vessels (Figure 1). This histopathological picture was described as consistent with the diagnosis of Gottron’s papules in the course of dermatomyositis or other disease from the lichenoid dermatitis group (Figure 1). The second histopathological biopsy, collected from a subcutaneous nodule from the elbow area, showed an inflammatory infiltrate, comprised mainly of lymphocytes and less numerous histocytes, penetrating to the subcutaneous tissue, surrounding blood vessels, nerves and located interstitially between collagen fibres and hyperplasia of fibrous connective tissue. There were no mucin deposits or signs of vasculitis observed (Figure 1). On the basis of the whole clinical picture and histopathological examination, the diagnosis of interstitial granulomatous dermatitis was made (Figure 1). Serum levels of muscle enzymes, tumour markers, thyroid stimulating hormone, anti-citrullinated protein antibodies and rheumatoid factor were within normal range. An indirect immunofluorescence assay showed antinuclear antibodies (ANA) titre of 1 : 320 with granular pattern of fluorescence; no antigen-specific antibodies were found.
The patient was administered methotrexate (15 mg) weekly and methylprednisolone at a starting dose of 16 mg daily with a gradual dose reduction of 4 mg monthly. This regimen was continued for 6 months without a significant change in the patient’s state. Currently the patient is receiving dapsone (25 mg and 50 mg, alternately) and no new skin changes have been observed.
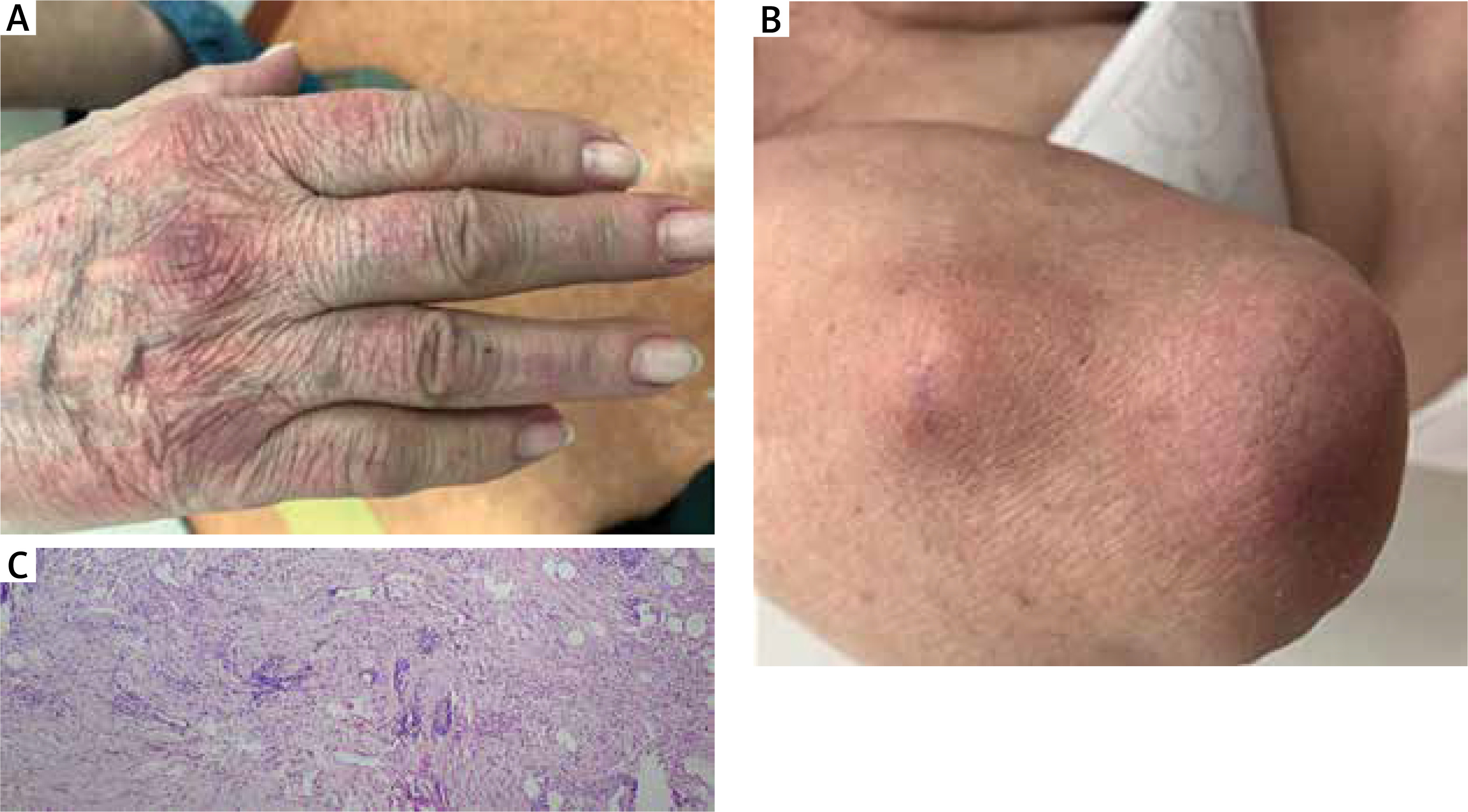
The other of described patients (case 2), a 70-year-old female was admitted to the Dermatology Department due to asymptomatic, violaceous skin patches overlying metacarpophalangeal joints, more prevalent on the dorsal region of the right hand in the area of the first and second metacarpophalangeal joint and erythema of the forehead, neckline and the skin of the left elbow; in this location, subcutaneous, hard, movable nodules were also present (Figure 2). Similarly to patient 1, there were no subjective symptoms associated with skin changes. First skin changes started to occur 2 years before the admission. The patient was suffering from arterial hypertension, hypercholesterolemia, glaucoma and cataract and was receiving long-term treatment with carvedilol, ramipril, indapamide, atorvastatin and acetylsalicylic acid. In order to confirm the diagnosis a biopsy of the subcutaneous nodule was collected from the right elbow area. In histopathological examination, perivascular and interstitial lymphohistiocytic infiltrates were described in the dermis (Figure 2). Although there were no changes in the epidermis, marked presence of fat tissue cells was observed directly below epidermis. The overall picture was described as consistent with diagnosis of IGD. An indirect immunofluorescence assay showed ANA titre of 1 : 320, a trace amount of anticytoplasmic antibodies was also detected. Autoantibody profile testing by immunodiffusion was negative. The patient was initially treated with topical glucocorticoids. Due to an unsatisfactory response to topical treatment, hydroxychloroquine (200 mg) was added, thanks to which a significant improvement was achieved. However, the complete resolution of the skin lesions was observed a few weeks after withdrawal of atorvastatin.
Figure 2
Clinical and histopathological presentation of case 2. A, B – asymptomatic, violaceous skin patches overlying small hand joints, resembling Gottron’s sign, with a subcutaneous, hard, movable nodule on the elbow. C – interstitial lymphohistiocytic infiltrate in the mid and deep dermis (H + E, 100×)
Interstitial granulomatous dermatitis, also described in the literature as Ackerman syndrome, is a rare cutaneous reaction pattern. It was presented in the literature by an American dermatopathologist A. Bernard Ackerman in 1993, although first cases of patients suffering from rheumatoid arthritis with the presence of asymptomatic skin papules with linear distribution were reported back in 1965 [1, 2]. Together with palisaded and neutrophilic granulomatous dermatitis and interstitial granulomatous drug reaction, this disease belongs to a group of reactive granulomatous dermatitis. However, some authors do not make a distinction between this condition and PGND due to some similarities in their clinical and histopathological patterns [3]. The disease occurs more frequently in females that in males, the average age of onset is 58 years. An exact pathomechanism of the disease is unknown, although a non-specific reaction of the autoimmune system is suggested as a possible cause on account of its frequent comorbidity with various autoimmune diseases. In approximately half of reported cases, there was IGD comorbidity with rheumatoid arthritis, although cases of concomitant systemic lupus erythematosus, autoimmune thyroiditis, autoimmune hepatitis, Churg-Strauss syndrome, Behçet’s disease, Lyme disease, lung coccidioidomycosis and various malignancies were described in the literature [1, 4]. Reported cases of interstitial granulomatous drug reaction were associated primarily with use of TNF-α inhibitors, β-blockers, calcium channel blockers, ACE inhibitors and furosemide. More recent reports have described the relationship between IGD and ledipasvir/sofosbuvir and topiramate [2, 5]. Cases related to IGD and statins have also been described, including simvastatin in a 68-year-old patient in whom it coexisted with elevated serum transaminases [6].
For many years the “classic” clinical presentation of the disease as described by Ackerman were thought to be erythematous patches, red or violaceous in colour, sometimes with symmetrical linear distribution on sides of the trunk. This manifestation, named the “burning rope sign” was considered to be pathognomonic of IGD, although subsequent reports have confirmed its prevalence in only 9% of patients [1]. More recent case reports, however, are highlighting the heterogeneity of the clinical picture of cases in which histopathological pictures suggest IGD. In the biggest up-to-date case study of 12 cases linked with a retrospective analysis of 53 previously described cases, the most common sign, occurring in 70% to 90% of patients was asymptomatic multiple erythematous papules, indurated plaques and infiltrative lesions [4]. Sometimes this characteristic clinical picture is absent and erythematous annular changes located most often on the sides of the neck, trunk and limbs develop instead. Lesions are typically asymptomatic. In our patients, the clinical picture was more like the one described in PGND, in which erythematous, violaceous papules and plaques covering extensor joints are often observed.
Considering a great variability of the clinical picture of IGD, its diagnosis requires a histopathological confirmation. A typical histopathological picture consists of infiltrate of epithelioid cells showing CD68 expression dispersed between bundles of collagen fibres in middle and deep layers of the skin. Histocytes may also surround necrobiotic collagen fibres, sometimes forming pseudorosettes and granulomas with solitary giant cells. In most cases of developed disease, degenerated collagen cells surrounded by histiocytes detach from the tissue, forming solitary islets, which is described as a “floating sign” [4]. In rare cases, disseminated eosinophilic and granulocytic infiltrates are present in the deeper layers of the skin, including multinucleated and atypical lymphocytes and plasma cells. Vasculitis and mucin should be absent. Epidermis is typically unchanged, except for cases of drug-induced reactive granulomatous dermatitis, in which vacuolization of the basal layer may be observed more frequently [1, 4].
Standards of treatment for IGD have not been well established yet, mostly due to rarity of the disease. To exclude a possible interstitial granulomatous drug reaction, careful analysis of all medications taken by the patient is advised as the withdrawal of the causative factor can resolve cutaneous lesions. Careful screening for underlying conditions is necessary in all cases of IGD. Majority of the cases described in the literature was treated with topical or systemic steroid therapy. Other immunosuppressive agents used in the treatment include methotrexate, cyclosporine A, intravenous IgG immunoglobulin, etanercept, tocilizumab and ustekinumab [7–10]. It is worth noting, however, that the main indication for systemic immunosuppressive treatment in most cases was the underlying rheumatic disease. There are also reports of successful treatment with dapsone and hydroxychloroquine, which has proven to be effective in our patients [7]. Approximately 20% of cases may resolve spontaneously.
In both of described cases, thorough laboratory and imagining studies have not revealed signs of active rheumatic, infectious or neoplastic disease. In histopathological examinations performed, distinct features of the drug reaction were not described. In both patients, changes confirmed histopathologically as IGD were manifesting clinically as asymptomatic, hard, movable, small nodules, located in subcutaneous tissue within the area of erythematous, violaceous lesions overlying joints. Such clinical picture resembling changes occurring in the course of dermatomyositis has not been previously described as characteristic for IGD.







