
Introduction
The neurofibromatoses are a group of three heterogeneous disorders including neurofibromatosis type 1 (NF1, Recklinghausen disease), neurofibromatosis type 2 (NF2) and schwannomatosis [1]. Recklinghausen disease is the most common of the three conditions.
Specific criteria for the diagnosis of NF-1 were created in 1987 [2]. Two of these seven clinical features need to be recognized for the positive diagnosis: six or more café-au-lait spots over 5 mm in greatest diameter in pre-pubertal individuals and over 15 mm in greatest diameter in post-pubertal individuals; two or more neurofibromas of any type or one plexiform neurofibroma; freckling in the axillary or inguinal regions; optic glioma; two or more Lisch nodules (pigmented iris hamartomas); distinctive osseous lesions such as sphenoid dysplasia or thinning of the long bone cortex with or without pseudarthrosis; a first-degree relative with NF-1 [2–4].
Recklinghausen disease is an autosomal dominant disorder affecting approximately 1 : 3000 individuals worldwide [5]. Neurofibromatosis type 1 is caused by inherited or de novo germline mutations in the NF1 tumour suppressor gene [6]. It is thought that somatic loss-of-function of the second allele results in the development of tumours such as cutaneous neurofibromas, plexiform neurofibromas and malignant peripheral nerve sheath tumours which are the most characteristic in the Recklinghausen disease [6]. The gene, located on chromosome 17, has one of the greatest frequencies of spontaneous mutation in the whole human genome [7, 8].
A major feature of NF1 is the development of localized cutaneous neurofibromas. Such types of lesions are manifested in > 99% of adults with NF-1 and are responsible for major negative effects on quality of life [5]. Therefore, patients with neurofibromatosis type 1 often identify the presence of cutaneous neurofibromas as their greatest burden [6]. So far the natural history of cutaneous neurofibroma formation has been poorly understood and despite growing knowledge about the role of NF-1 gene mutations in the pathogenesis of Recklinghausen disease, the molecular diagnostics of this pathology is still difficult and its molecular basis requires further studies [6, 8]. The protein product of the NF-1 gene is neurofibromin, it is expressed in all cells however the highest expression levels are present in neurons, Schwann cells, glial cells, melanocytes and leukocytes [7, 8]. The neurofibromin belongs to the group of neoplasm suppressor proteins and its main role it to inactivate the Ras protein, which is the membrane protein that controls intracellular signalling networks and participates in processes like differentiation and proliferation [7, 8]. Histologically neurofibromas consist of Schwann cells, perineural cells, fibroblasts and a collagenous matrix; also they contain an infiltrating immune cell population that is comprised of macrophages and mast cells [9, 10].
Currently, there is no effective way of treatment for patients with neurofibromatosis type 1. One method is surgical excision, however removing all lesions is usually not feasible due to the abundance of NF-1 and disease progression [5, 11]. Surgery is definitely needed when malignancy is suspected. Also surgical operations are indicated when tumours pressure other organs or make moving difficult or cause neurological symptoms like severe pain, epilepsy, and hydrocephaly [12]. Neurofibromatosis type 1 is considered as a benign tumour condition; however, malignant transformation has been reported in 2% of patients with NF1 or 4.2% of those older than 21 years of age [13]. Other techniques of tumour removal that can be offered to patients with NF-1 include less invasive surgical methods or conservative treatment. These less invasive ways of therapy are laser-based cutting, electrodessication, radiofrequency ablation and photodynamic therapy [14–16]. Topically applied treatments that can be put on cutaneous neurofibromas are examples of conservative therapy (drugs like rapamycin, imiquimod, liquid diclofenac or selumetinib) [6]. However, so far results of topically applied treatment have not been really efficient and satisfactory [6].
Aim
The aim of this paper is to present the clinical picture of patients with NF-1 and to report authors’ own observations related to their surgical treatment.
Material and methods
A retrospective study aiming to present a group of patients with Recklinghausen disease was planned. Medical data of all the patients with NF-1, who had been treated in our clinic between 1992 and 2017, were carefully studied. The study was carried out in 39 patients with neurofibromatosis type 1 diagnosed in the Department of Plastic, Reconstructive and Aesthetic Surgery and our plastic surgery outpatient clinic. Each patient underwent a clinical examination, and photographic documentation was carried out. A thorough medical history of each patient was also recorded. This way, a patient database was created on the basis of the medical data, ambulatory cards, hospital histories, and photos. All the patients were otherwise healthy and did not have any other congenital anomaly. The collected data included age, sex, family history, parts of body affected with neurofibromas, and methods of treatment.
On the basis of the patients’ documentation and database created, a retrospective analysis of treatment was conducted. Informed, written consent has been obtained from patients. The protocol of the study was approved by the ethics review board of the Medical University of Lodz, and the study was conducted according to the Declaration of Helsinki principles. The Ethics Committee of the Medical University of Lodz has approved this study.
Results
There were 14 men and 25 women in the examined group. The patient’s age, at the time of the first consultation, was from 11 months to 61 years of age (the average age was 30 years and 2 months).
All patients had cutaneous neurofibromas localized in different parts of the body. The three most frequent localizations were the abdomen, back, and face (Figure 1). Other body parts affected by skin nodules included the scalp, neck, buttocks, upper and lower limbs also with hands and feet. All patients had multiple lesions, more than 10 neurofibromas were detected, between 5 mm to 3 cm in diameter. Additionally in 15 patients, apart from small cutaneous nodules, a giant neurofibroma was recognized.
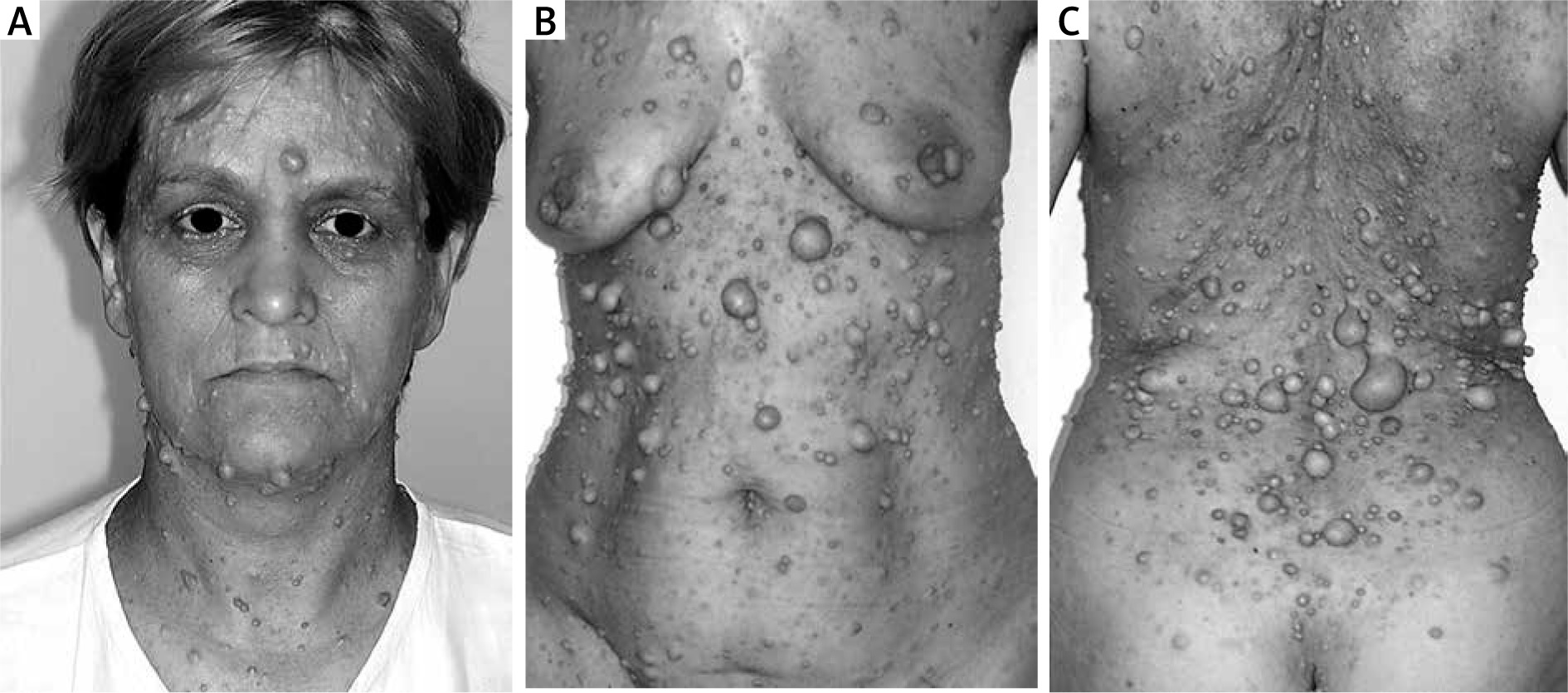
All patients had discomfort due to the presence of cutaneous nodules especially because of their visibility and multiplicity. Some patients also noted pain, mainly those who had a giant lesion in the area of the face or limbs or when they observed tumour growing. The patient who had limb neurofibroma additionally observed oedema and paraesthesia.
Surgical treatment was performed in 31 patients (10 with giant tumours and 21 with smaller nodules). Those with giant tumours were operated in general anaesthesia, those with cutaneous neurofibroma – in local anaesthesia. In patients with giant neoplasm partial resections of neurofibroma mass were carried out. Only in 1 patient the whole tumour was resected in one-stage procedure. In the remaining patients with giant tumours, neurofibromas were too extensive to perform only one excision, in 1 patient surgical procedures were done even 6 times. In patients with typical tumours, surgical excision with primary closure or local flap procedures were introduced. In one intervention maximum 6 lesions were excised. Multiplicity of lesions made it impossible to remove all lesions at once. In 15 (71.4%) patients there were more than 11 operations, in 3 (14.3%) less than 5, and in remaining 3 (14.3%) between 6 to 10. All patients were satisfied with the treatment, and majority of them declared their wish to continue surgeries. In 8 patients from the analysed group surgical interventions were not performed. One of them was operated on in another medical centre, one child will be planned for surgery when it is older, and 6 patients did not agree to offered treatment.
Clinical and physical improvement was observed in all patients, mainly due to reduced quantity of nodules and/or mass reduction of giant tumour (Figure 2). Nevertheless the aesthetic result was not always good, especially in patients with giant neurofibromas, however here extensiveness of neoplasm was wide and multi-stage procedures were necessary (Figure 3).
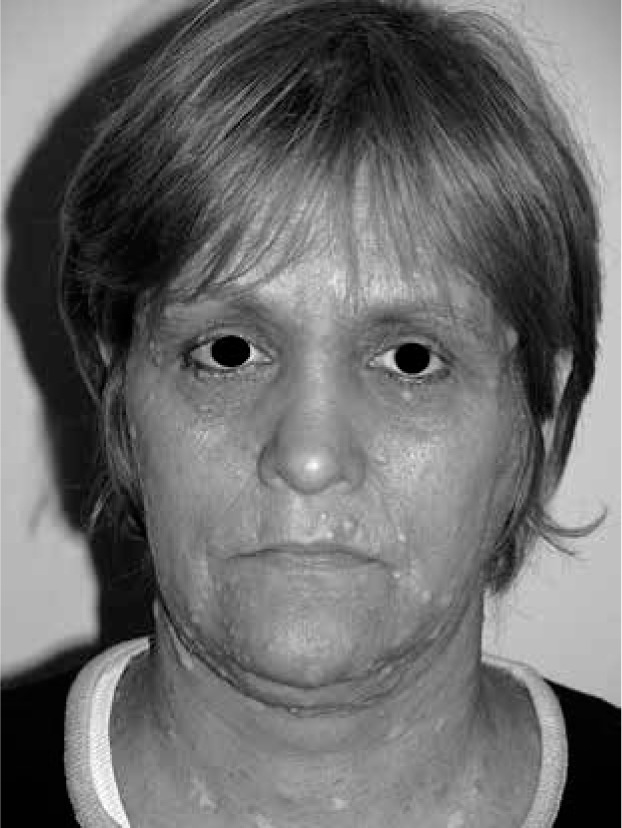
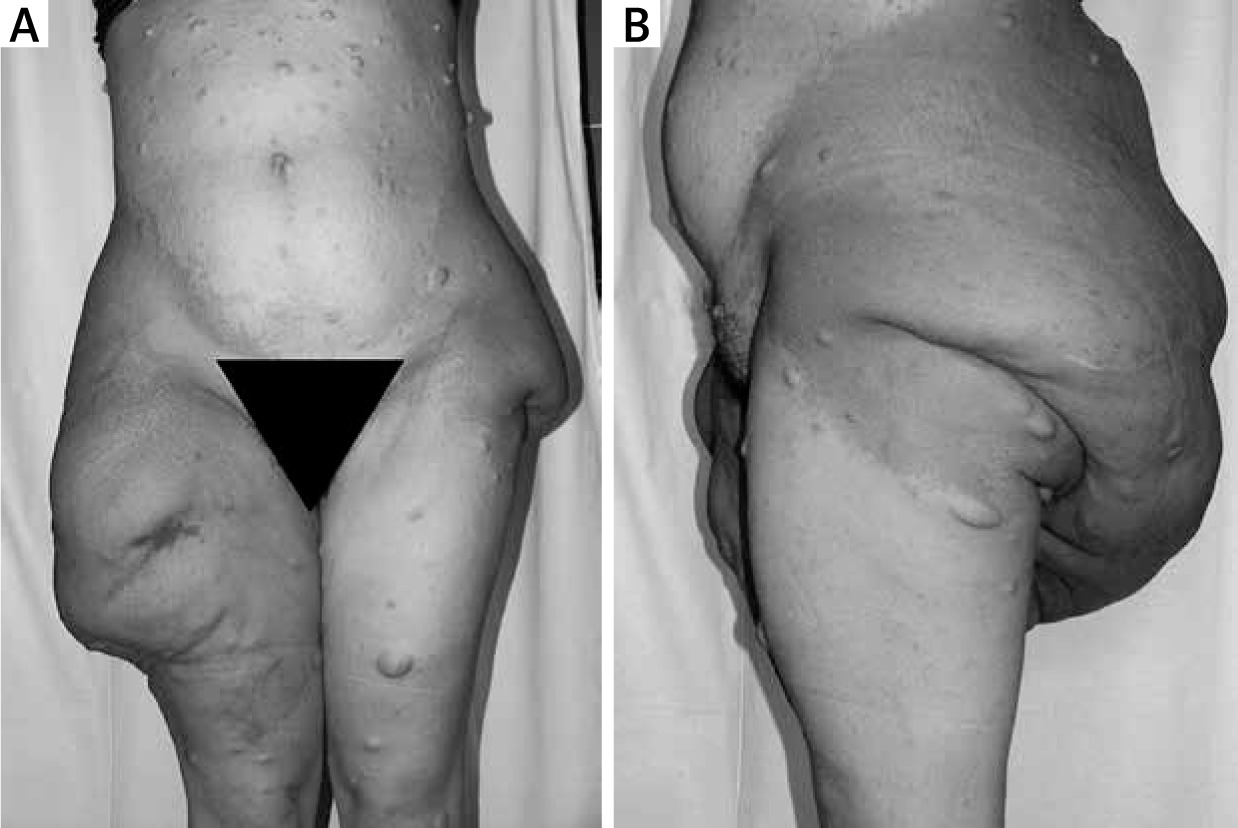
Figure 3
Patient with NF-1 with multiple skin neurofibromas and giant tumour in the area of buttocks and thighs
In 17 (43.5%) patients the family history was positive; NF-1 was present in a first-degree relative.
Discussion
Patients with Recklinghausen disease present with a diverse spectrum of manifestations. The clinical picture can vary and may include skin lesions (skin-fold freckling, café au lait macules, tumours of peripheral nervous system), neoplasm in the central nervous system (like pilocytic astrocytoma), disorders in eyesight called Lisch nodules (iris hamartoma), bones abnormalities (mainly scoliosis) and rarely malformations in the cardiovascular system (pulmonary stenosis and renal artery stenosis) [2–4, 6, 7]. In this report we focused on cutaneous neurofibromas present in patients with NF-1 and their surgical removal which represents a challenge to surgeons in patients with thousands of tumours.
Treatment of patients with Recklinghausen disease is mainly symptomatic. The main aim is early detection and excision of neurofibromas, especially when they are small and one-stage surgical procedure is possible. It is also important to perform surgery when mass of neurofibroma is getting bigger and daily activities are becoming too difficult for the patient [3]. Authors indicated that cutaneous nodules in NF-1 appear around puberty, increase with age, and undergo periods of rapid growth in puberty and pregnancy [5, 17, 18]. The presence of multiple tumours or giant neoplasm causes different complaints in patients. They suffer mainly from pain, paraesthesia, oedema and deformations in the body area affected by neoplasm, which can lead to significant morbidity and influence negatively quality of life [5, 17, 18]. Similar concerns were noticed in our group of patients. Good functional and aesthetic results after operations in our patients were achieved as most of them declared their wish to continue surgeries.
Bieniek et al. paid attention to the fact that in most patients with NF-1, due to disease progression, treatment cannot be recognized as finished. Authors advise to consider further surgeries when the lesions are growing and new neurofibromas appear [12]. We agree with the above-mentioned authors. Our patients are still under care of our outpatient clinic and are qualified for next operations.
The literature notes that in 30–50% of patients with Recklinghausen disease it is impossible to state familial occurrence of neurofibromatosis type 1, and symptoms of the disorder are related to mutation de novo [3, 13]. Similar data come from our analysis, since only in 43.5% of patients we confirmed familial occurrence of NF-1. More detailed genetic tests seem to be necessary in patients with neurofibromatosis type 1 to evaluate more precisely the frequency of familial occurrence.
Conclusions
Due to the polymorphism of the clinical picture and unpredictable course of the disease, there is no one standard of treatment of patients with Recklinghausen disease. Excision of tumours in the early stage of their development gives better functional and aesthetic results. Due to disease progression, surgical procedures that are undertaken are usually multi-stage ones. When introduction of operation is delayed, usually the excision of the whole mass of neurofibroma is not possible, which causes danger of tumour re-growing and cancer transformation.







