
Introduction
Asthma is one of the most common chronic respiratory diseases, with severe public health implications for children and adults. Approximately, 300 million people worldwide have asthma, and it is likely that by 2025 a further 100 million may be affected [1]. Despite many scientific advances, the burden of asthma emerged as a global public health problem. Only in the United States, 14 million working-days are lost each year and asthma-related socioeconomic costs reach almost 56 million US dollars [2]. The prevalence of asthma has significantly increased over the last decades, with large variations in different areas. Incidences of asthma are much more common in high-income countries than rural regions [3]. There is no single cause of asthma. The following interactions between environmental exposures, genetic susceptibility, and specific host factors lead to the development of this complex and multifactorial disorder [2]. Although underlying mechanisms of asthma are not fully understood yet, the suspected symptoms may include control of respiratory bronchial hyperactivity and chronic lower airway inflammation that is characterized by cough, wheezing, chest tightness, and shortness of breath. Advances in research on asthma not only depend on better recognition of the disease, but also integrated cross-disciplinary approaches and scientific tools which will enable us to provide a new insight into the complex pathogenesis of asthma, involving interactions between environmental and genetic factors [3]. Numerous complex mechanisms also involve various regulatory T cell subsets and Toll-like receptors. It is suspected that this kind of pathway could derive, to some extent, from alteration in microbiota composition caused by multiple lifestyle factors. The “hygiene hypothesis” was the first to imply a link between microbes and allergy by suggesting that childhood exposure to particular microorganisms contributes to the development of the immune system. The original hygiene hypothesis was expanded and formulated taking into consideration current lifestyle changes, like rise in antibiotic use, the prevalence of caesarean birth and milk formula feeding, and finally changes in modern dietary patterns which have profound consequences for the intestinal microbiome composition [4].
The healthy human microbiome contains as many as 1014 bacteria among 1000 bacterial species which is comparable to the number of cells of a single human [5]. The gut microbiota of adults is predominantly colonized by Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria. Aerobic bacteria like Lactobacilli, Enterobacteriaceae, and Streptococci species colonize mainly the duodenum, proximal small intestine, and stomach, while anaerobes including Bifidobacterium, Bacteroides, Clostridium, Prevotellaceae or Lachnospiraceae dominate the colon and distal small intestine [6]. The composition of the human gut microbiome evolves toward an adult-like diversity 3 years after birth; during this time it is incessantly subjected to multiple exposures that influence its ecology. Many environmental factors, especially those related to diverse diet and the place of residence characterized by microbial-rich environments (e.g., on a farm) have been inversely associated with childhood asthma [7]. It is suspected that human gut microbiota composition has an impact on the maturation of the host immune cells. Any disturbance in the initial development of the microbiota may cause many immunologic disorders like allergy and asthma, and thereby may be recognized as a crucial environmental factor linked to the health-to-disease transition [8]. Recently, several studies have revealed that early life dysbiosis of the gut microbiota precedes an altered risk of asthma [9–12]. However, it is still unclear if dysbiosis within the gut can actually initiate disease-promoting mechanisms or just reflects associated phenomena, such as altered patterns of immune response taking into account microbes and environmental factors.
Over the last several years, our knowledge in the field of human gut microbiota has expanded and allowed us to understand its crucial role in maintaining human health [13]. Clinical studies indicate that gut microbiota plays an important role in affecting asthma development [11]. It is suspected that there is a “critical window” of gut colonization in early infancy; during this period shaping of immune maturation takes place and contributes to higher susceptibility to allergic airway inflammation. Although the exact mechanisms that determine the influence of gut microbiota on the “gut–lung axis” and immune maturation remain unclear, they certainly include immune cell differentiation and production of metabolites. Taken together, these findings suggest an association between microbial communities and host immune development as well as allergic airway inflammation development [13].
The novel tools made a profound difference in the analysis of genes and genomes within overall microbial communities and offered innovative options for the limitations and obstacles related to classic culture-based approaches. One of the most powerful approaches to investigating complex gut microbiota composition is represented by Next-Generation Sequencing technology (NGS) [14]. Metagenomic profiling by 16S rRNA high-throughput sequencing and gene amplicon analysis was applied in some recent studies concerning on role of gut microbiota alteration in the development of asthma [15].
Aim
The aim of this study was to analyse the nature of human gut microbiota patterns among adult patients with asthma in comparison to healthy controls.
Material and methods
Study design
The study was based on a case-control design to analyse the faecal microbiota of adults with asthma. Twenty samples were obtained, 13 from patients with asthma and 7 from healthy people for a control group. Patients were not taking antibiotics or probiotics for at least 30 days prior to study entry. In the conducted survey, none of the patients qualified for the study reported episodes of allergic rhinitis, food allergies, and dust mite allergy. Additionally, none of them have ever experienced hypersensitivity reactions to non-steroidal anti-inflammatory drugs. Unfortunately, we did not have the results of skin prick tests and total IgE level for all participants, therefore it was decided not to include this parameter in the analysis of the obtained results. The clinical characteristics of the patients and the members of the control group are summarized in Table 1. All the patients in the study group were treated on a long-term basis for asthma, therefore, asthma control was assessed. In 10 cases, control status according to the Global Initiative for Asthma (GINA) guidelines was “controlled asthma” and for 3 patients it was “partly controlled asthma”. Detailed information about asthma treatment among the study group is summarized in Supplementary Table S1. Faecal samples of about 4 g were collected into sterile tubes. Samples were immediately frozen at –80°C and stored until further analysis. The samples were blinded; the researchers did not have access to patients’ data. Total bacterial genomic DNA was extracted from the faecal samples with the Genomic Mini AX Stool Spin (A&A Biotechnology, Gdansk, Poland) according to the manufacturer’s instructions. The isolated DNA was then stored in the freezer at –20°C. In the next step, the isolated DNA served as a template in the PCR amplification of the 16S rRNA.
Table 1
Baseline characteristics of participants
The NGS library preparation was based on PCR amplification of the V3 hypervariable region of the 16S rRNA gene. The amplification reaction was performed using the Kapa HiFi Master Mix polymerase (Roche, Rotkreuz, Switzerland). After amplification, the amplicons were analysed in 10% polyacrylamide gel electrophoresis. The amplicons were purified with the NucleoSpin gDNA Clean-up XS Kit (Macherey – Nagel, Allentown, PA, USA), the DNA concentration was assessed using the compact fluorometer Qubit 2.0 (Invitrogen, Waltham, MA, USA). For each sample, a library was prepared according to the Meyer and Kircher protocol excluding the fragmentation and enrichment step [16]. Prepared libraries were sequenced on the MiSeq 2500 (Illumina, San Diego, CA, USA) device with the set of reagents for paired reads with 250 cycles. The primary analysis of the obtained sequences was performed, consisting of the demultiplexing and FASTQ file generation with the MiSeq Reporter software (Illumina). The forward and reverse reads (R1 and R2) of each sample were exported from the MiSeq system for analysis in the FASTQ.GZ file. Adapters and low-quality bases and sequences were removed with cutadapt. Mothur was used to process the sequence data and to perform taxonomic assignments following the Operational Taxonomic Unit (OTU) approach from the MiSeq Standard Operating Procedure [17, 18]. In the first step, the forward and reverse reads were merged. Reads exhibiting any ambiguous positions were subsequently removed. Next, reads were aligned to the SILVA reference database [19]. Sequences from chloroplasts, mitochondria, archaea, eukaryotes, and unknown were deleted before the OTU clustering at 97% identity. The resulting reads are screened for the presence of chimeras using UCHIME [20]. Finally, sequences are clustered into OTUs.
Biodiversity indices and principle statistics analyses on taxonomic profiles were analysed in MicrobiomeAnalyst [21]. The participants were classified into the following two groups: (1) n = 6, control; and (2) n = 12, asthma. During statistical analysis of the results, to obtain statistically significant results, it was decided to discard the results of samples No. 11 (asthma group) and No. 20 (control group), which differed a lot from the other samples. Alpha diversity was performed using the phyloseq package. Furthermore, Principle Coordinate Analysis (PCoA) using the Bray-Curtis distance-based method was applied, and the statistical significance of the clustering pattern in ordination plots was evaluated using Permutational ANOVA (PERMANOVA). The level of similarity between samples was measured by Bray-Curtis distance.
The study was approved by the Bioethical Committee of the Medical University of Lodz. Written informed consent was obtained from all the participants.
Statistical analysis
NGS sequences, besides Mothur, were also identified at the bacterial phyla using Kraken with the custom Kraken database. Data were analysed within the Galaxy platform, with a Galaxy Web server instance supported by the National Microbiology Laboratory, Public Health Agency of Canada (PHAC NML Galaxy). The Mothur-based method was useful to identify a diverse bacterial microbiota from the RNA-seq datasets, but it was not able to classify any of the bacterial sequences further than the genus level. After analysing the microbiome data using the Kraken software, the number of OTU counts for each sample was converted to a percentage (100% – total number of counts in each sample) and used for statistical tests. Non-parametric statistical analyses were performed using the software OriginPro, Version 2019 (OriginLab Corporation, Northampton, MA, USA). The Kruskal-Wallis ANOVA and Mann-Whitney tests were used to compare the above two groups of subjects. The statistical differences were considered significant at the p < 0.05 level.
Analyses of differential/relative abundance features (of metagenome data) were done using STAMP (Statistical Analysis of Metagenomic Profiles) software package, version 2.1.3. The differential abundance between the samples was calculated using a one-sided G-test (w/Yates’) + Fisher’s test with asymptotic-CC confidence intervals (0.95) and Storey FDR multiple test correction for two samples analysis in STAMP.
Bioinformatics and statistical analyses of the sequences were performed on Linux (Ubuntu 18.04, 64-bit) operating system, installed on a Dell server equipped with 24 CPU cores and 124 GB RAM.
Results
The composition of the gut microbiota among the study and control groups was examined. Presented data demonstrated different abundances of certain bacteria in the gut microbiota of patients with asthma compared with a healthy control group, and that the gut dysbiosis in patients with asthma may be associated with the aberrant faecal microbiota composition. To better illustrate the differences in the intestinal microbiota composition in the study and control groups, only the most numerous taxa are presented in the graphs. We have presented the profiles of intestinal microbial communities for asthma and control groups and individual patients at a genus (Figure 1) and species (Figure 2) taxonomic level.
Figure 1
The detailed view of individual differences between asthma and control groups at the genus level
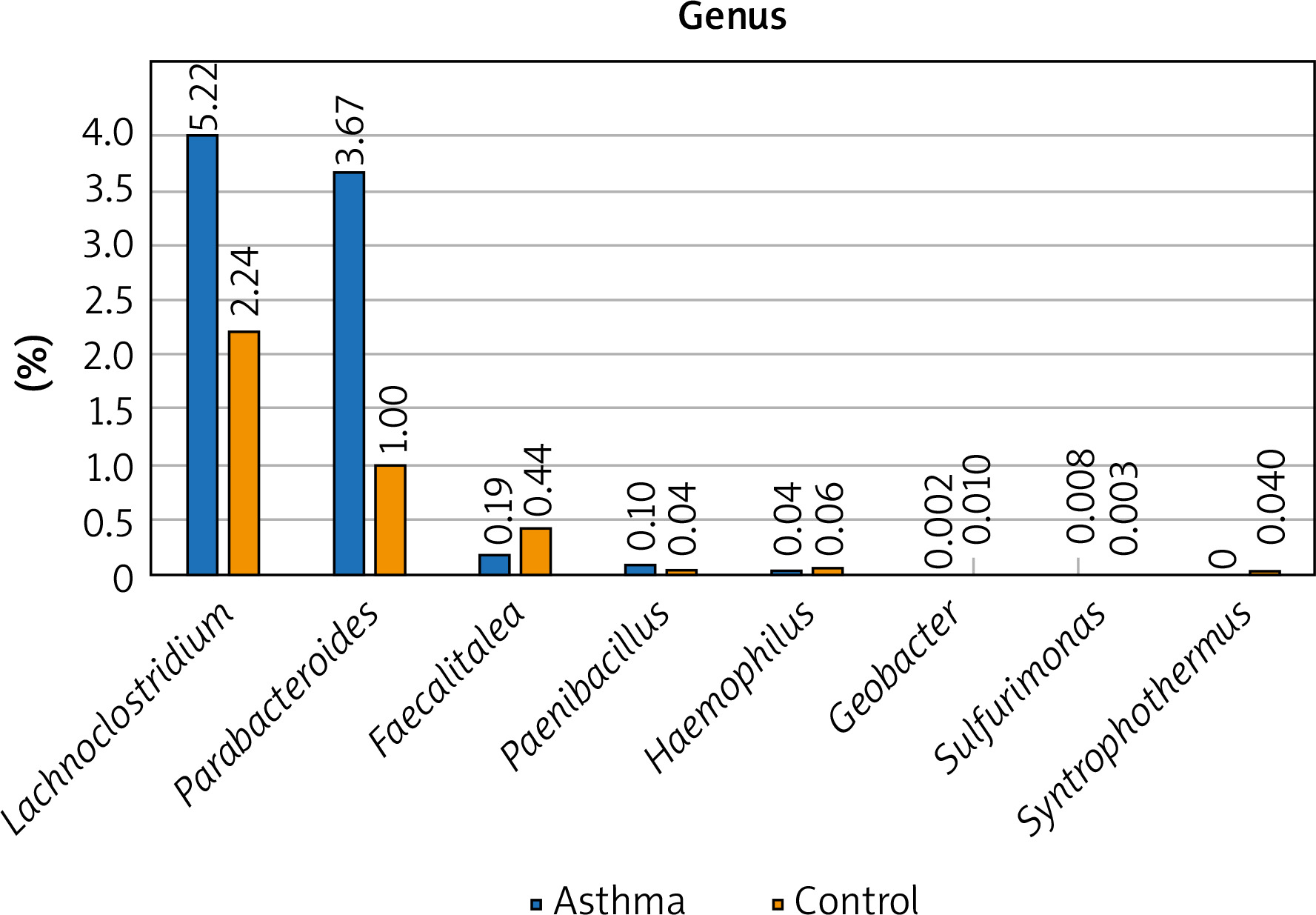
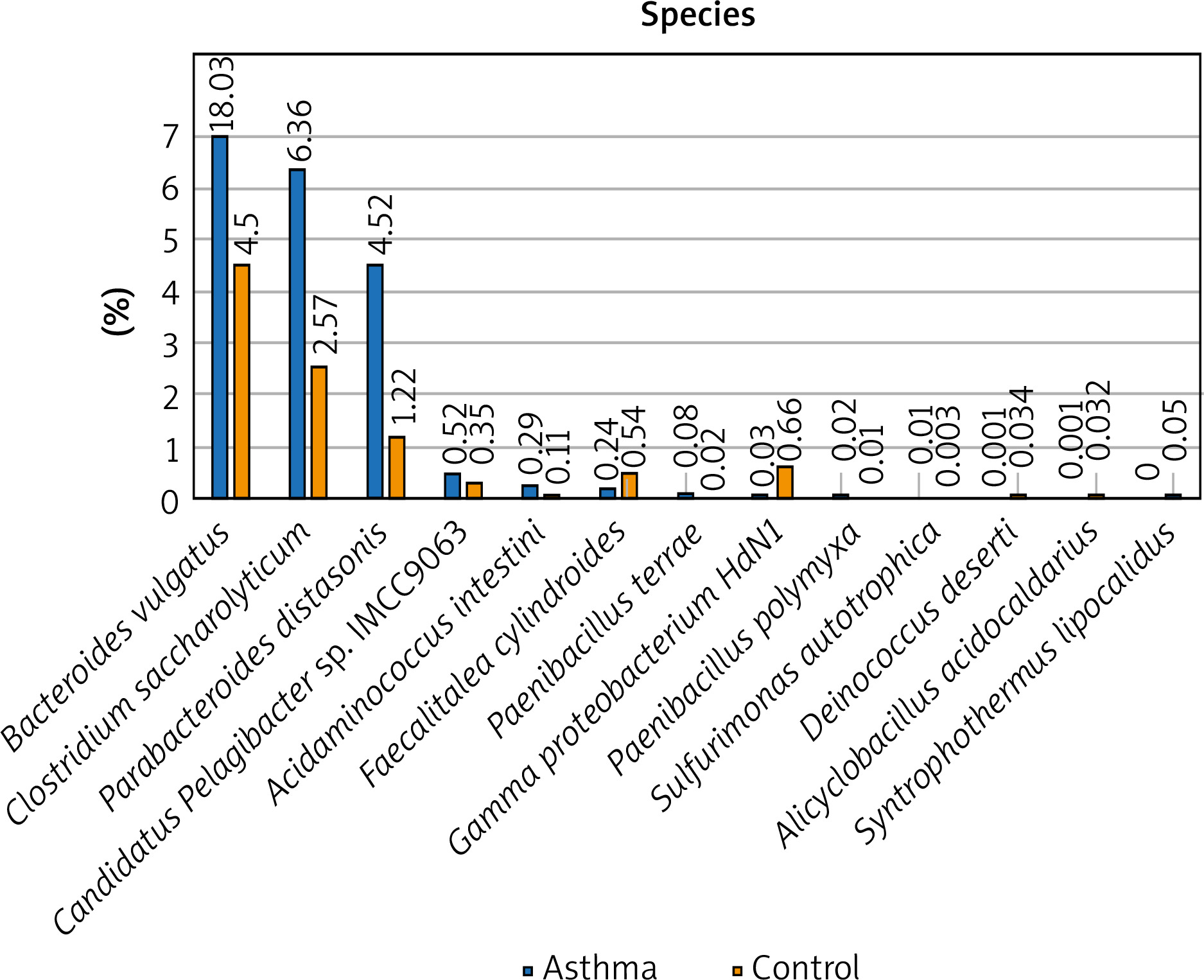
Figure 2
The detailed view of individual differences between asthma and control groups at the species level
At the genus level, Prevotella, Ruminococcus, Bacteroides and Oscillibacter constitute the 4 most numerous genera of bacteria constituting nearly 55% of all bacteria among asthma and control groups. The number of Bacteroides in the asthma group is twice as high as in the control group, while in the case of Prevotellaceae it is 3 times smaller. Similarly, the number of Ruminococcaceae and Oscillospiraceae is much higher among the control group, however, the difference was not statistically significant (p > 0.05). Differential taxon abundances between asthma patients and healthy subjects at the genus level for the 20 most abundant genus were presented in Supplementary Figures S1 A, B. Analysis shows statistically significant differences between the asthma group and the control group in the case of Faecalitalea (p = 0.02459), Parabacteroides (p = 0.00369), Haemophilus (p = 0.01633), Paenibacillus (p = 0.03935), Syntrophothermus (p = 0.00203), Methanocella (p = 0.02385), Geobacter (p = 0.00641), Sulfurimonas (p = 0.03184) and Lachnoclostridium (p = 0.0492) (Figure 1).
At the species level, Bacteroides vulgatus, Oscillibacter valericigenes, Bacteroides thetaiotaomicron, and Prevotella dentalis are the 4 most numerous species of bacteria constituting 35% of all bacteria among the asthma group and 29% among the control group, respectively. The number of Bacteroides vulgatus in the asthma group is four times higher than in the control group and most importantly, this difference is statistically significant (p < 0.05). The second species of the Bacteroides genus – Bacteroides thetaiotaomicron, is also predominant in asthma patients. On the other hand, the number of Oscillibacter valericigenes and Prevotella spp. is much higher among the control group, however, the difference was not statistically significant (p > 0.05). Differential taxon abundances between patients and healthy subjects at the species level for the 20 most abundant species was presented in Supplementary Figures S2 A, B. Analysis shows statistically significant differences between the asthma group and the control group in case of Bacteroides vulgatus (p = 0.0492), Clostridium saccharolyticum (p = 0.0492), Parabacteroides distasonis (p = 0.00273), Gamma proteobacterium HdN1 (p = 0.01586), Faecalitalea cylindroides (p = 0.02459), Candidatus Pelagibacter sp. IMCC9063 (p = 0.0492), Acidaminococcus intestini (p = 0.01798), Syntrophothermus lipocalidus (p = 0.00203), Alicyclobacillus acidocaldarius (p = 0.00641), Deinococcus deserti (p = 0.03974), Paenibacillus terrae (p = 0.01468), Sulfurimonas autotrophica (p = 0.04026) and Paenibacillus polymyxa (p = 0.04687) (Figure 2).
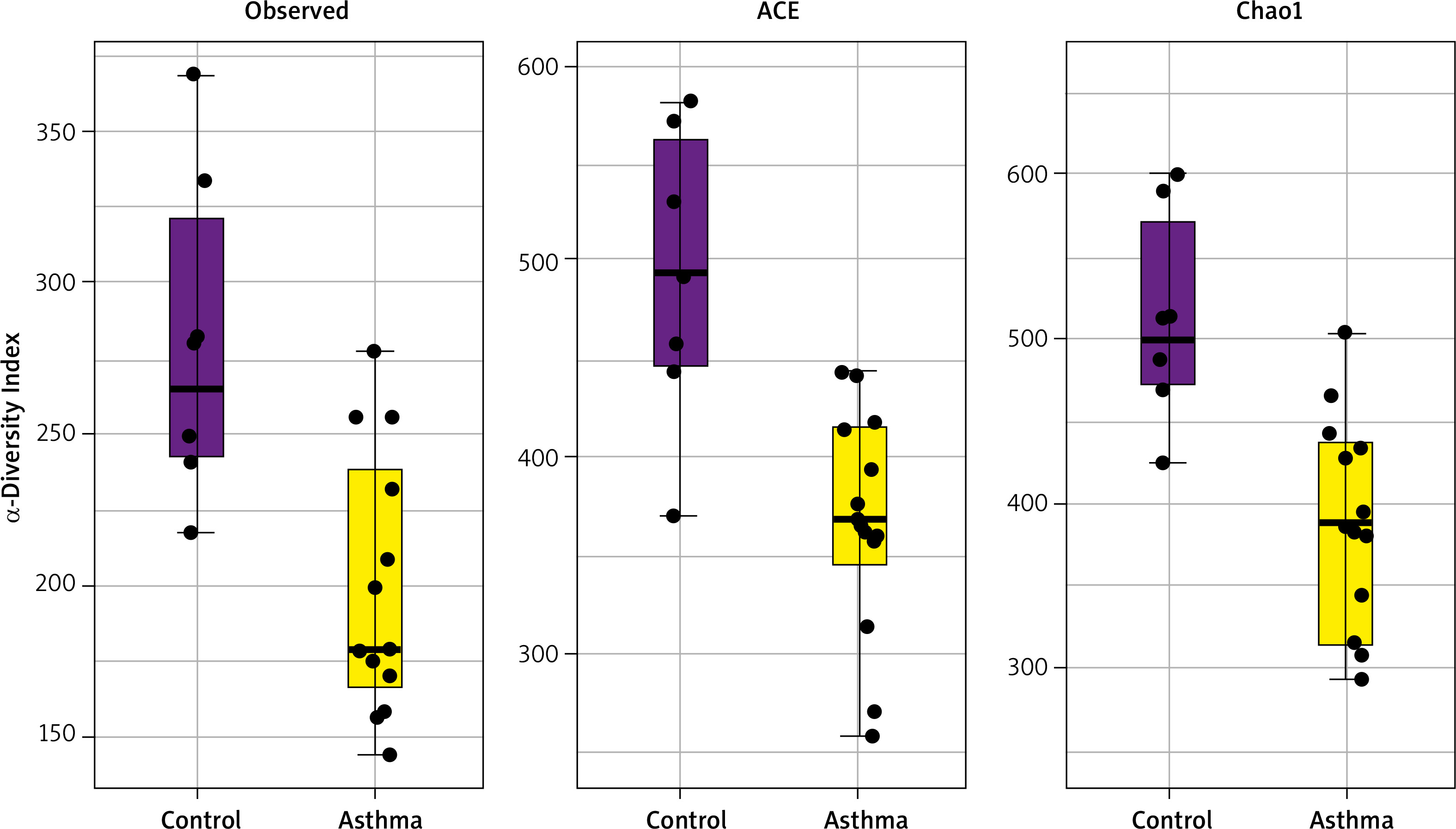
Alpha diversity analyses are plotted across samples and reviewed as box plots for each group (Figure 3). Within-sample diversities of the asthma patients and healthy controls are significantly different (p < 0.05). Observed, ACE and Chao1 diversity were significantly decreased in the asthma group compared to the control group (pObserved = 0.015898; pACE = 0.011102; pChao1 = 0.0044047). The median α diversity of gut microbiota measure across groups is lower in asthma patients than in the control group’s value. Supplementary Figure S3 shows the alpha diversity measure across all the samples.
Figure 3
α-diversity index of gut samples, measured by Observed, ACE and Chao1. Notes: α-diversity index of gut samples, measured by Observed, ACE and Chao1, is plotted for patients with asthma (yellow) and controls (purple). The line inside the box represents the median, while the whiskers represent the lowest and highest values within the 1.5 interquartile range. Outliers as well as individual sample values are shown as dots
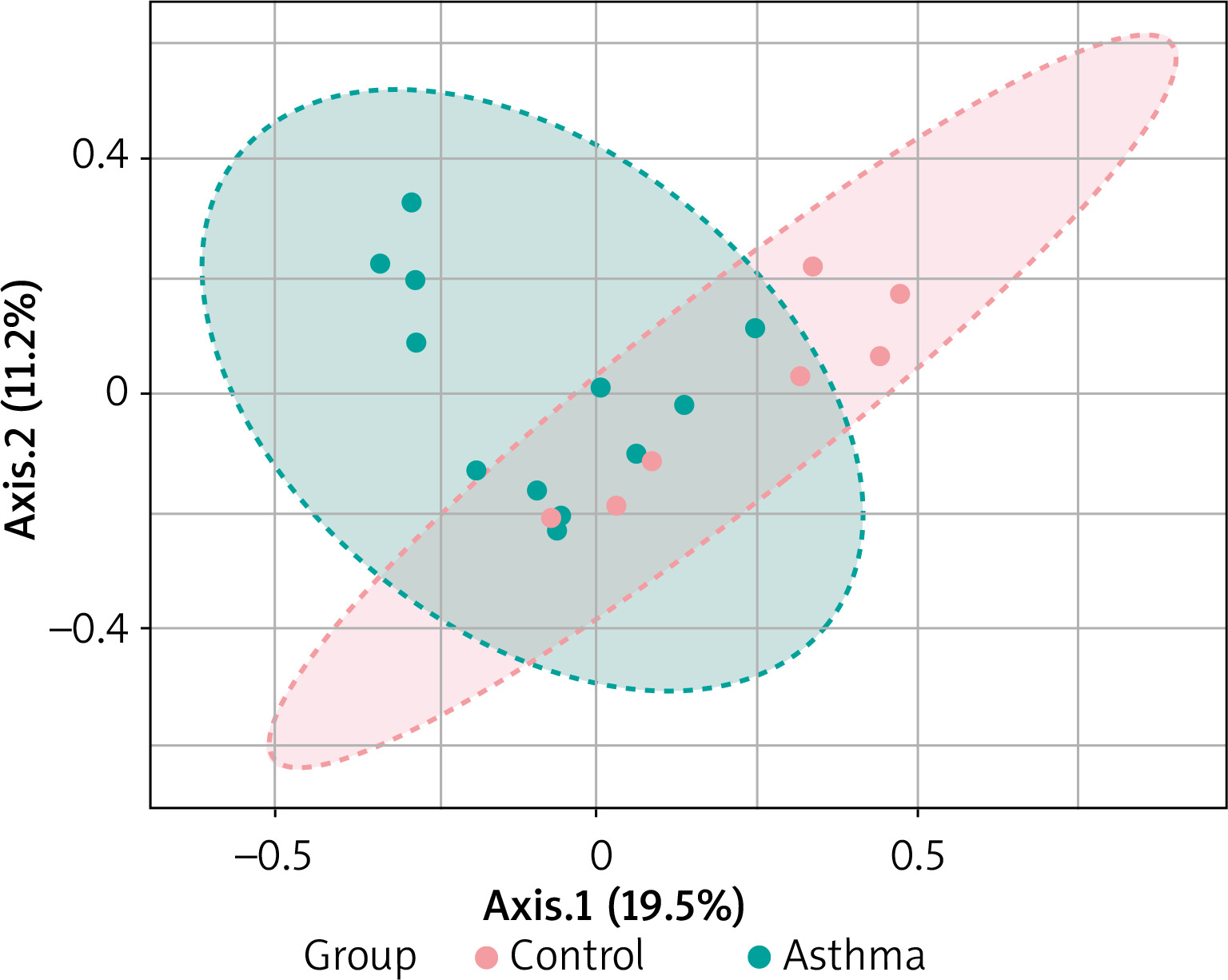
Beta diversity assessment of gut microbiome was performed using the phyloseq package. Principles Component Analysis (PCoA) was used, and a scatterplot was generated to determine the phylogenetic distances between bacterial communities. The axes indicate the percentages of variation in the data for the bacterial communities. Principal coordinate analysis (Figure 4) showed that gut community compositions of asthma are widely dispersed in contrast to the tight clustering observed for the control group (F-value: 1.792; R2 = 0.10072; p-value < 0.013).
Figure 4
2D PCoA plot for β-diversity analysis. Notes: The explained variances are shown in brackets. Dot symbols in different representing colours, clustered together are circled in red (control) and light blue (asthma), respectively
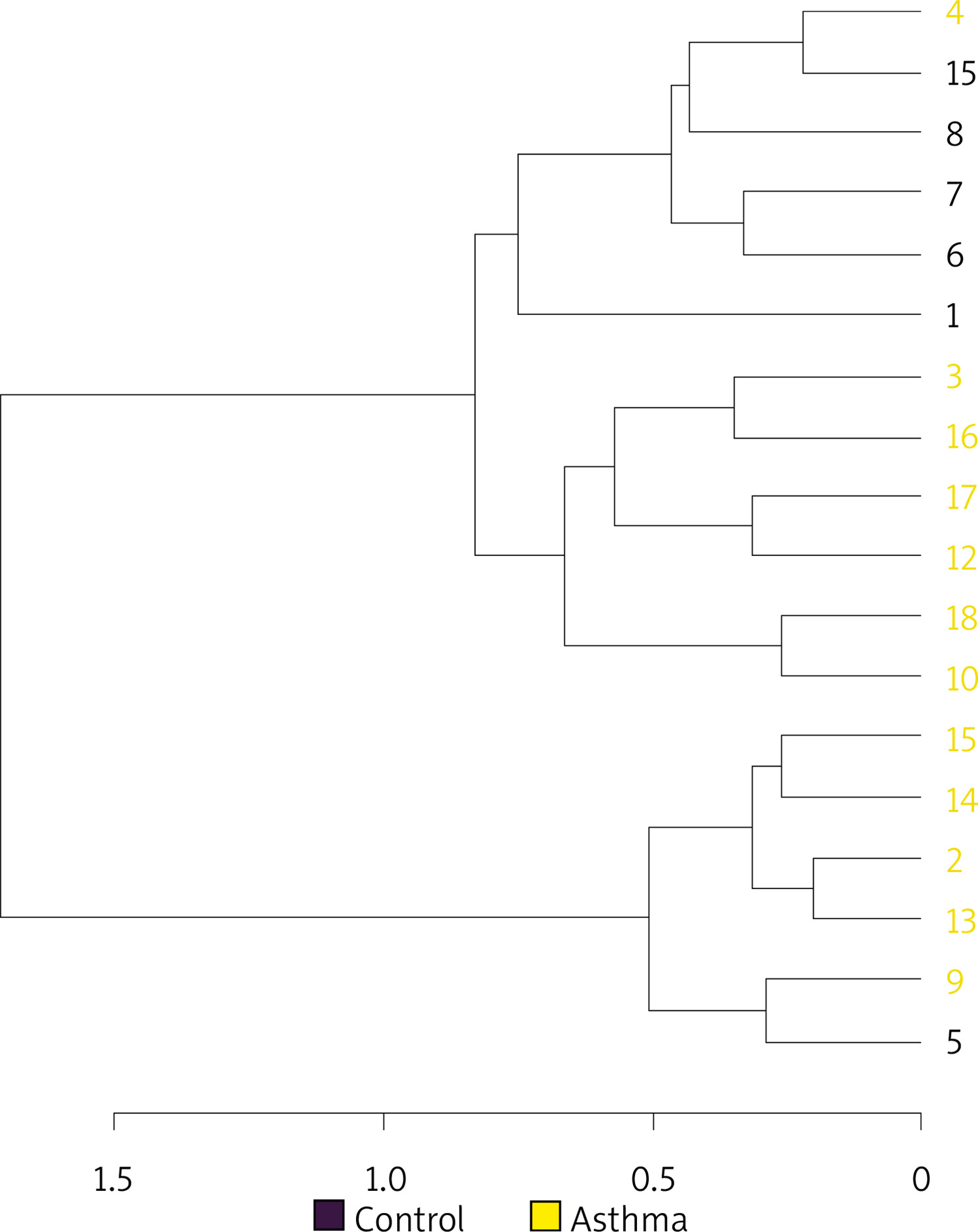
Hierarchical cluster analysis results are shown in Figure 5 as a dendrogram. Similarity index results were lower in the inter-group comparison than in the intra-group comparison indicating a change in the composition of gut flora in asthma patients.
Discussion
Bacteria within the gut are responsible for numerous functions such as the production of vitamins, food fermentation, absorption of ions, and enhanced immune functions. Recent research has demonstrated that the human gut microbiota composition can confer either significant health benefits or increase susceptibility to particular disorders [22, 23]. Especially, a microbial imbalance has been associated with the development of several allergic diseases and asthma [6]. The influence of microbiota on the formation and development of asthma is a widely discussed topic. We compared the composition of the gut microbiome in asthma patients and healthy volunteers during our research. The study revealed significant differences in the human gut microbiome composition between asthma patients and the healthy control group, which were present at each tested level (genus and species). The presented study has some limitations concerning mainly the number of patients. There are several related studies involving a larger number of participants, however, it can be partly compensated by the use of modern culture-independent technique, which allows for comprehensive, sensitive, specific, and very accurate analysis while providing high-quality evidence [9, 14, 24, 25]. Additionally, a precise selection of study participants minimizes the impact of the mentioned factor on the study outcomes. Statistical analysis of received data enabled the identification of several genera and species of the gut microbiota, a number of which differed significantly between the study group and the control group.
The study group had a significantly greater number of as many as 4 different genera of bacteria such as Parabacteroides, Paenibacillus, Sulfurimonas, and Lachnoclostridium, while the control group has a greater population of Faecalitalea, Haemophilus, Syntrophothermus, Methanocella and Geobacter. In previous related studies, the early life abundance of the bacterial genera Lachnospira, Veillonella, Faecalibacterium, and Rothia was significantly decreased. This dysbiosis had an additional effect on the reduction in the faecal acetate level and dysregulation of enterohepatic metabolites [11]. In addition, a lower relative abundance of genus Bifidobacteria, Akkermansia, and Faecalibacterium resulted in the highest risk of asthma among neonates [12]. During their research, Stiemsma et al. linked asthma at preschool age with gut bacterial dysbiosis, particularly with reduction in Lachnospira in favour of Clostridium spp. in the first 3 months of life [26]. These conclusions were confirmed in the next study in which the decrease in Veillonella, Faecalibacterium, and Rothia as well as the increase in Lachnospira resulted in numerous asthma incidences [27]. Moreover, the amount of Clostridium spp. is closely related to the increased risk of asthma in several other studies [9, 24, 25]. Interestingly, a higher abundance of Bifidobacteria and Lactobacillus was detected in the gut of breast-fed infants, whereas formula-fed infants’ gut microbiome was characterized by a greater proportion of Bacteroides, Streptococcus, Clostridium, Enterobacteriaceae, and Veillonella [28]. Additionally, breast-feeding is associated with decreased risks of asthma compared to formula-fed infants [29]. The composition of gut microbiota is a complex issue that can be affected by numerous factors which, in turn, may adversely affect the study [3–11]. Therefore we have taken steps to minimize this probability through the appropriate qualification of patients for the study, taking into account numerous risk factors like antibiotic and probiotic administration, age, sex, body mass index, or smoking status. Interestingly, there was no effect of smoking on the abundance of presented genera and species among the study and control groups.
If we look at particular species of bacteria, the amount of Bacteroides vulgatus, Clostridium saccharolyticum, Parabacteroides distasonis, Candidatus Pelagibacter sp. IMCC9063, Acidaminococcus intestini, Paenibacillus terrae, Sulfurimonas autotrophica, and Paenibacillus polymyxa were significantly higher in patients with asthma than in the controls. On the other hand, the relative abundance of Gamma proteobacterium HdN1, Faecalitalea cylindroides, Syntrophothermus lipocalidus, Alicyclobacillus acidocaldarius, and Deinococcus deserti was negatively associated with asthma occurrence. Researchers investigated that Clostridium difficile colonization at 1 month of age was associated with asthma incidence at the age 6 to 7 years [9]. Current research suggests that gut microbiota can influence immune response even at distant sites, like lungs, via multiple mechanisms. For example, the gut of adult asthma patients is more abundant in histamine secreting bacteria, compared to healthy volunteers [30]. Ferstl et al. suspect that histamine induces protective responses against asthma histamine 2 receptors in the lungs [31]. What is more, some studies indicated that specific Clostridia strains are able to induce expansion of Treg cells in the colonic mucosa as well as reduced IgE production among OVA-sensitized, pathogen-free BALB/c mice [32]. Similarly, in another study, BALB/c mice treated with Limosilactobacillus reuteri were characterized by expansion of Treg cells in the circulation and significantly reduced inflammatory response to OVA sensitization [33].
Compared to healthy individuals, bacterial diversity was significantly lowered among the asthma group, which is evidence of gut microbiota depletion in asthma patients. What is more, analysis of β diversity showed that the gut community compositions of asthma are widely dispersed in contrast to the tight clustering observed for control. Finally, the similarity index was found to be lower in the inter-group comparison than in the intra-group comparison, which confirmed changes in gut microbial composition among the asthma group. Many researchers obtained consistent results that asthmatic patients had lower airway and gut microbiome diversity compared to healthy controls [27–29]. The importance of microbiome diversity is shown by studies connecting lower gut microbiome diversity measured during the first week of life with the occurrence of asthma at school age [9]. Furthermore, low gut microbiota diversity during the first month of life was associated with asthma in children at the age of 7. Early gut microbial diversity turned out to be very important for asthma development but did not apply to the other allergic manifestations [10].
Conclusions
Our research revealed significant differences in the human gut microbiome composition between asthma patients and the healthy control group, which is consistent with the previous studies in this topic. The link between gut microbiota and autoimmune diseases has attracted attention of many researchers and studies have been conducted on this association in certain diseases, including asthma. Nevertheless, the knowledge gap concerning the detailed relationship between gut microbiota composition and asthma remains unclear. Despite some limitations, the study offers new, potentially useful information and provides an excellent basis for further research. Because of gut microbiota complexity and dynamic environment influenced by multiple factors, further studies with a larger number of well-characterized patients and controls are necessary to study the exact role of the gut microbiome in the course of asthma. Additionally, further research should focus on the diverse mechanisms of action used by gut microbiota to alleviate the course of asthma as well as on the potential use of microbiota-directed therapies in the treatment of asthma.







