
Juvenile xanthogranuloma (JXG) is a rare condition which belongs to a group of histiocytoses. It is also described as the most common non-Langerhans cell histiocytosis (non-LCH). Considering the large variability of its clinical presentation, histopathological verification of cutaneous lesions is necessary. The management of JXG involves observation, surgical treatment or systemic treatment. We present a case of a 2.5-month-old male patient with diffuse skin lesions in course of JXG, present since birth.
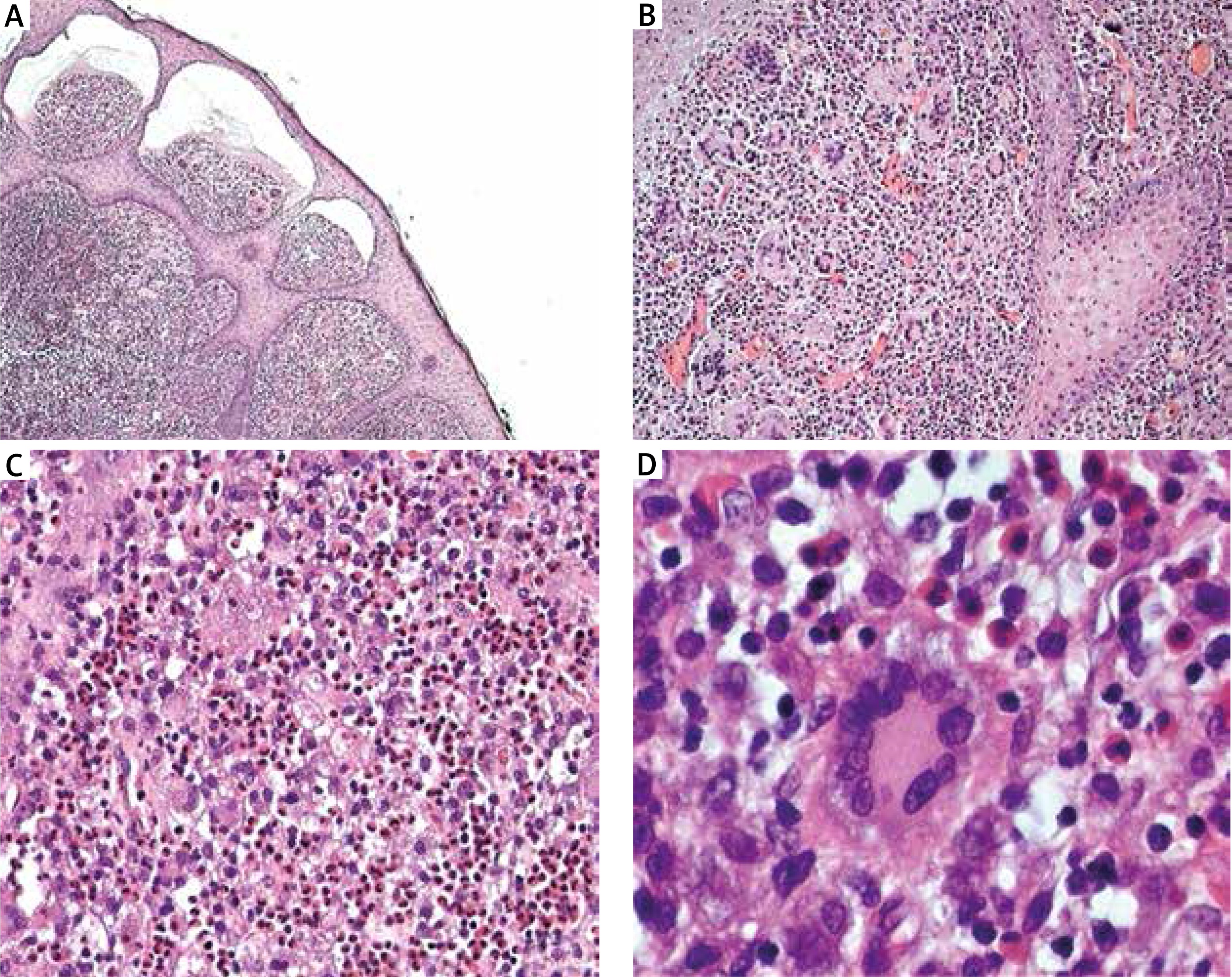
The 2.5-month-old male infant presenting yellow-orange skin nodules localized on his upper and lower limbs and trunk, was admitted to the Department of Dermatology. Lesions have been already present since the child’s birth and have become gradually larger. New nodules have also appeared. The boy was born on time, with perinatal history burdened with posthypoxic status and paralysis of the facial nerve, born by Caesarean section, and scored 10 points in the Apgar scale. The child was not treated for any other diseases before the hospitalization. He was vaccinated according to the valid calendar but the cycle of vaccinations was interrupted because of skin lesions occurrence. Physical examination at the admission demonstrated presence of numerous yellow-orange, dense nodules and papules (Figures 1 A–C). Skin lesions were localized on the right forearm, in the area of the right iliac ala, on the second finger of the left hand, on the third toe of the left foot and on the sole of the right foot. Basic laboratory blood tests conducted during hospitalisation revealed minor macrocytosis (90.7 fl), minor monocytosis (1.38 × 103/μl) and a slightly extended activated partial thromboplastin time (37.7 s). Other laboratory tests were normal. The infant had ophthalmological and neurological consultations. The examinations did not reveal any pathological symptoms. There were no abnormalities in abdominal ultrasonography and ultrasound examination of soft tissues within skin nodules demonstrated the typical presentation for JXG. The largest nodule located close to the right iliac ala was excised for histopathological examination with the shave biopsy method. The lesion manifested microscopically as a dense histiocytic proliferation in the papillary and reticular dermis. Epidermis was thin with focally elongated rete ridges (Figure 2 A). Foamy histiocytes and giant cells were dominating in the infiltration. Some of the giant cells were the Touton-type (Figure 2 B). Touton giant cells (xanthelasmic giant cells) were characterized by a ring of multiple nuclei surrounding a central, homogenous, eosinophilic cytoplasm, while a foamy, pale cytoplasm surrounded the nuclei (Figure 2 C). Among foamy histiocytes, abundant eosinophils were also observed. There were no mitotic figures present (Figure 2 D). On the basis of microscopic examination and clinical presentation, the diagnosis of JXG was confirmed. The patient remains under supervision of our dermatology outpatient clinic. During the follow-up visit after one month, regression of majority of dermatological lesions was observed (Figures 1 D–F).
Figure 1
Skin lesions on the right forearm, in the area of the right iliac ala and on the third toe of the left foot at the admission to the Department of Dermatology (A–C). The same skin lesions 1 month later (D–F)
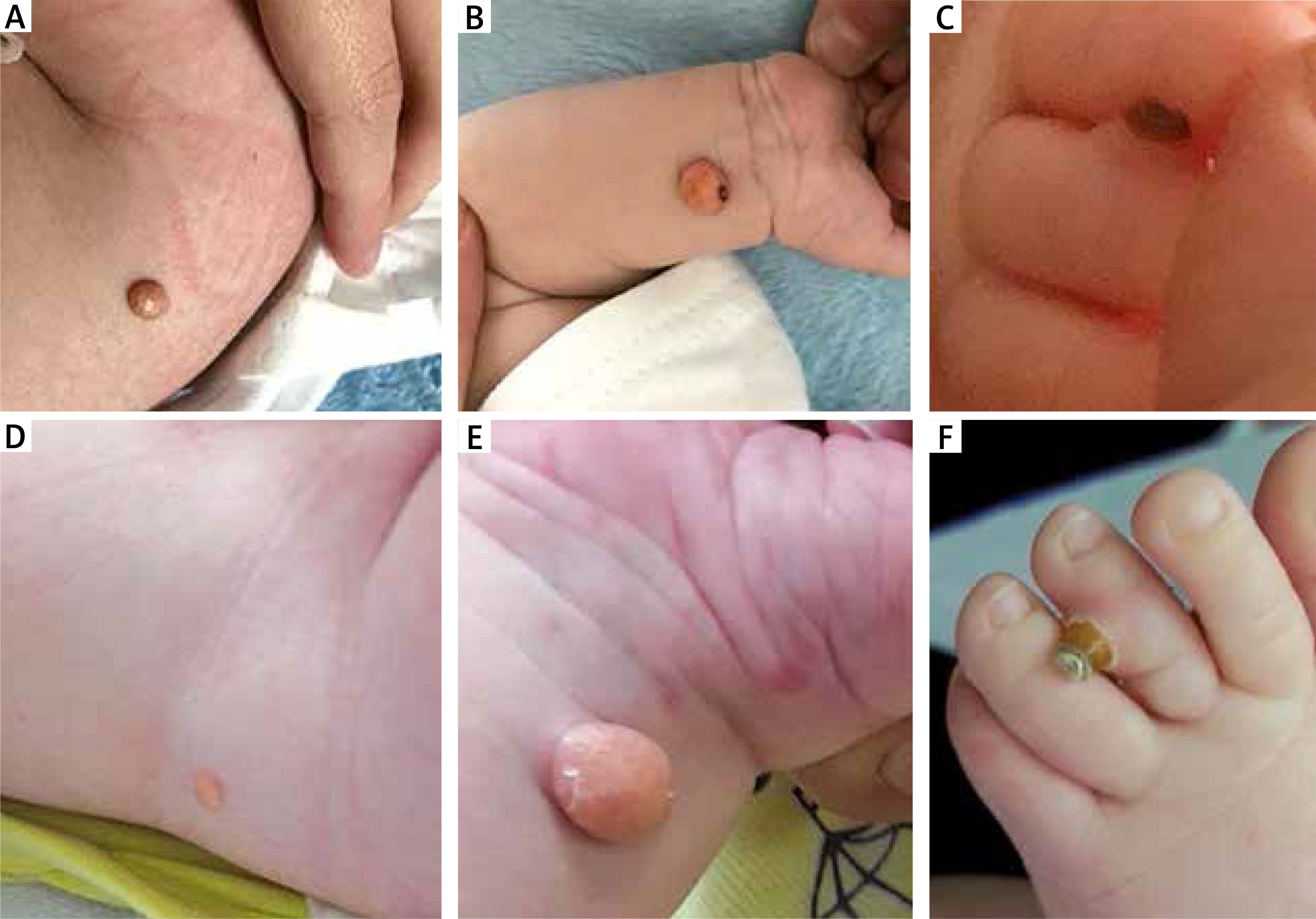
Figure 2
A – Histopathological examination of a skin sample taken from the right iliac ala, H + E, magn. 4×. Dense histiocytic proliferation in the papillary and reticular dermis. Epidermis lining the skin thin, with focally elongated rete ridges. B – HE, magn. 10×. Foamy histiocytes and giant cells dominating in the infiltrate. Some giant cells are of the Touton giant cell type (giant cells seen in lesions with high lipid content). C – Microscopic slide, HE, magn. 10–20×. Abundant eosinophils among foamy histiocytes. No mitotic figures. D – H + E, magn. 40×. Touton giant cell (xanthelasmic giant cell) with a characteristic ring of multiple nuclei surrounding a central homogenous eosinophilic cytoplasm, while a foamy, pale cytoplasm surrounds the nuclei
Juvenile xanthogranuloma is a rare condition belonging to the group of histiocytoses and the most common non-Langerhans cell histiocytosis (non-LCH). The condition belongs to the group of histiocytoses associated with proliferation of dendritic cells. It was first described by Adamson in 1905 [1]. Later, in 1912, McDonagh described 5 new cases of the disease [2]. In 1950s, Helwig and Hackney described 140 cases of the condition and gave it its name: xanthogranuloma or juvenile xanthogranuloma, used ever since [3].
Dermatological lesions secondary to JXG occur often in children and youth, and rarely in adults. Most commonly skin lesions develop in the first year of life [4]. Sixty-four percent of dermatological lesions develop before the age of 7 months and 85% before the age of one year. In approximately 10% of patients, skin lesions may be present since birth [5] as in the presented case. Younger age favours more diffuse forms of the disease [4]. Male infants are affected slightly more commonly than female ones and no sex predilection is observed in adults [4]. The prevalence of the disease is approximately 1 per million but in fact it may be much higher as single dermatological lesions may be misdiagnosed [6, 7].
The aetiology of the disease is not understood. It is assumed that abnormal response of macrophages to infection or trauma may be the cause of this condition [5].
Skin lesions often have the form of a single yellow-orange nodule, 1 cm in diameter or less, commonly as diffuse lesions occurring within the scalp, neck or extremities and trunk [4]. Non-typical presentations as plaque-like lesions are rarely observed but have been reported as well [8].
The disease may involve other internal organs, including lungs, kidneys, heart, central nervous system, adrenal glands, pituitary gland, bones and bone marrow. However, the most common non-dermal localisation of pathological lesions is the eyeball [5]. Eye may be involved even in patients with no skin manifestation of the disease. On the other hand, in patients with the dermatological form of JXG eyeball lesions is present in approximately 0.5% of cases [5, 9]. In case of patients who do not have dermatological symptoms, the diagnostic and therapeutic process is extremely complicated as an ocular malignant neoplasm has to be considered in differential diagnosis. Ocular lesions sometimes require systemic treatment because they may lead to cataract or blindness [9].
The JXG is also associated with neurofibromatosis type 1 (NF1) and with juvenile myelomonocytic leukaemia (JMML) [7]. Dermal lesions usually precede or develop at the time of leukaemia diagnosis. Patients with NF1 and JXG are also at increased risk of JMML [7]. For that reason, blood cell count with manual smear should be performed in monitored patients, especially in those with coexisting NF1.
The diagnosis of JXG is mostly based on clinical presentation of skin lesions and histopathological examination is essential for diagnosis confirmation. Dermoscopy proves to be useful in diagnostics of the disease, just like in overall dermatological diagnostics. Dermoscopic image of a lesion in polarized light demonstrates a yellow structureless pattern of a nodule with peripheral erythematous border. This presentation is called the “setting sun sign” [10]. Ultrasound examination is another non-invasive method applicable to verification of the diagnosis. Ultrasound examination with a linear transducer shows characteristic absence of posterior enhancement and Colour Doppler does not demonstrate any pathological flows within lesions [11]. In the discussed patient, the ultrasound presentation of examined skin nodules suggested JXG. Non-invasive techniques may be helpful in the assessment and monitoring of patients whose lesions had not been verified histopathologically.
Histopathological examination may demonstrate presence of numerous histiocytes, foamy cells and Touton giant cells [12]. It is recommended to avoid misdiagnosis of a malignant neoplastic process in a child. However, the excision biopsy is not required, and shaved biopsy is usually performed in order to save a patient from an excessive surgical procedure. Differential diagnosis considers other dermatological entities, such as dermatofibroma, mastocytoma or Spitz nevi [4]. Immunohistochemical tests are recommended in differentiation Langerhans cell histiocytosis (LCH) from JXG. In case of JXG, positive reactions of CD68, factor XIIIa and anti-CD4 are usually observed. S-100 protein is usually negative as that marker is typical of LCH [13, 14].
Dermatological lesions in course of JXG do not require treatment but only a regular, periodic observation as they often disappear spontaneously. Sometimes skin lesions require surgical removal because of aesthetic conditions, especially within the face or scalp. Patients require ophthalmological monitoring for approximately 6 months. Systemic treatment may be considered in case of internal organs involvement. Glucocorticosteroids, Vinblastine and Methotrexate are most commonly used [4].
The presented case demonstrates that skin lesions in patients with diagnosed JXG may have a variable clinical presentation, ranging from single to diffuse skin lesions, also present from the birth. The diagnosis requires histopathological confirmation to avoid misdiagnosis of malignant disease. Considering possible development of lesions in internal organs, paediatric, ophthalmological and neurological consultations are necessary as well as imaging diagnostics. Systemic treatment is rarely used. Majority of patients presenting lesions limited to the skin requires only a strict dermatological observation. Physicians should remember about the disease to avoid exposure of a paediatric patient to unnecessary excision of dermal lesions.







