
Introduction
Autoimmune blistering disorders (AIBD) are characterized by the development of circulating autoantibodies directed to desmosomal proteins in the pemphigus group and to basement membrane zone (BMZ) proteins in the pemphigoid group of disorders [1]. Both groups contain several entities with varying course, therapy and prognosis, therefore the establishing of the proper diagnosis is crucial. Therefore it is mandatory to find characteristic immunological features in patients’ biopsy and sera [2, 3].
How to properly take and collect material for diagnostic tests?
Direct immunofluorescence
The first step of the diagnosis is direct immunofluorescence (DIF) which is the gold standard if AIBD is suspected. The biopsy should be taken from the normal, perilesional skin or mucosa, never from the bulla or erosion. It is recommended to take a punch biopsy (3–4 mm) and at a distance of 1-2 cm from the lesion. From mucous membranes (especially oral mucosa) clipping of healthy mucosa should be taken using rounded scissors. It is very important that the tissue is not physically damaged during harvesting, e.g. crushed with tweezers. Tissues should be taken under anaesthesia with lidocaine in aerosol, avoiding injections since such trauma of mucous membrane may lead to epithelium separation and its losing during transportation, which may lead to false negative test results. Moreover, mucous membrane does not require sewing after taking a biopsy since it heals very easy after bleeding stops. Tissue sample should be transported in Michel’s solution or 0.9% saline if the transportation time is less than 24 h. In case the procedure cannot be performed properly, a patient should be referred to a referential dermatology department for consultation and collection of all necessary examinations [4, 5].
Characterization of target antigens
For the characterization of target antigens, it is necessary to take the patient’s blood (5–10 ml without anticoagulant). After centrifugation patient’s serum should be sent in ambient temperature to the laboratory experienced in AIBD diagnostics and stored in the freezer until study [4, 5].
Pemphigus group
Pemphigus vulgaris
Pemphigus vulgaris (PV) usually starts with blisters and erosions in the oral cavity (mucous membrane pemphigus), then skin of any area may be affected (mucocutaneous pemphigus). In both subtypes DIF shows IgG deposits in intercellular spaces (ICS) of epithelium/epidermis, in some cases C3 is also present (Figure 1 A) [6]. To finally diagnose PV, it is necessary to characterize circulating autoantibodies using at least one of currently available methods: indirect immunofluorescence (IIF), ELISA, BIOCHIP, or immunoblot [7]. Routinely, IIF is performed as the first using two preferable substrates – monkey oesophagus and guinea pig oesophagus being the source of desmoglein 3 (Dsg3) and 1 (Dsg1), respectively (Figures 1 B, C) [8]. Subsequently, BIOCHIP or ELISA containing recombinant Dsg 3 and Dsg1 should be performed [9]. In the mucosal subtype, autoantibodies react with Dsg3 exclusively, whereas in the mucocutaneous variant autoantibodies react with both Dsg3 and Dsg1. Additionally, ELISA allows to determine the exact concentration of antibodies in the patient’s serum, which is used in the treatment monitoring [10]. Recent studies showed that the sensitivity and specificity of BIOCHIP and ELISA in pemphigus diagnostics and type differentiation are comparable [11]. Both BIOCHIP and the recently used multiparametric ELISA allow for quick, one-step and simultaneous identification of autoantibodies directed against various antigens in several sera, which is particularly useful in patients with an unusual clinical picture and convenient for use in small laboratories [12, 13]. Routinely, immunoblot and immunoprecipitation are rarely used [14]. Histopathology is not necessary for diagnosis but may be helpful in differentiating it from other types of pemphigus such as IgA pemphigus or pemphigus vegetans. In case of PV suprabasal acantholysis with a sparse inflammatory infiltrate is observed [15].
Figure 1
Immunofluorescence studies: A – PV – IgG deposits in intercellular spaces of epidermis, B – PV – circulating IgG antibodies in intercellular spaces of human oesophagus, C – circulating IgG antibodies in intercellular spaces of guinea pig oesophagus in PV, D – reactivity of circulating IgG antibodies with rat bladder in PNP, E – circulating IgG antibodies in the roof of salt split skin in BP, F – circulating IgG antibodies in the floor of salt split skin observed in antiepiligrin cicatricial pemphigoid, anti-p200 pemphigoid and EBA, G – BP – positive reaction of circulating IgG antibodies with NC16A domain of BP180 antigen on ELISA, H – BIOCHIP mosaics – positive reaction of circulating IgG antibodies with BP180 (NC16A) typically for BP, I – granular IgA deposits in dermal papillae in DH
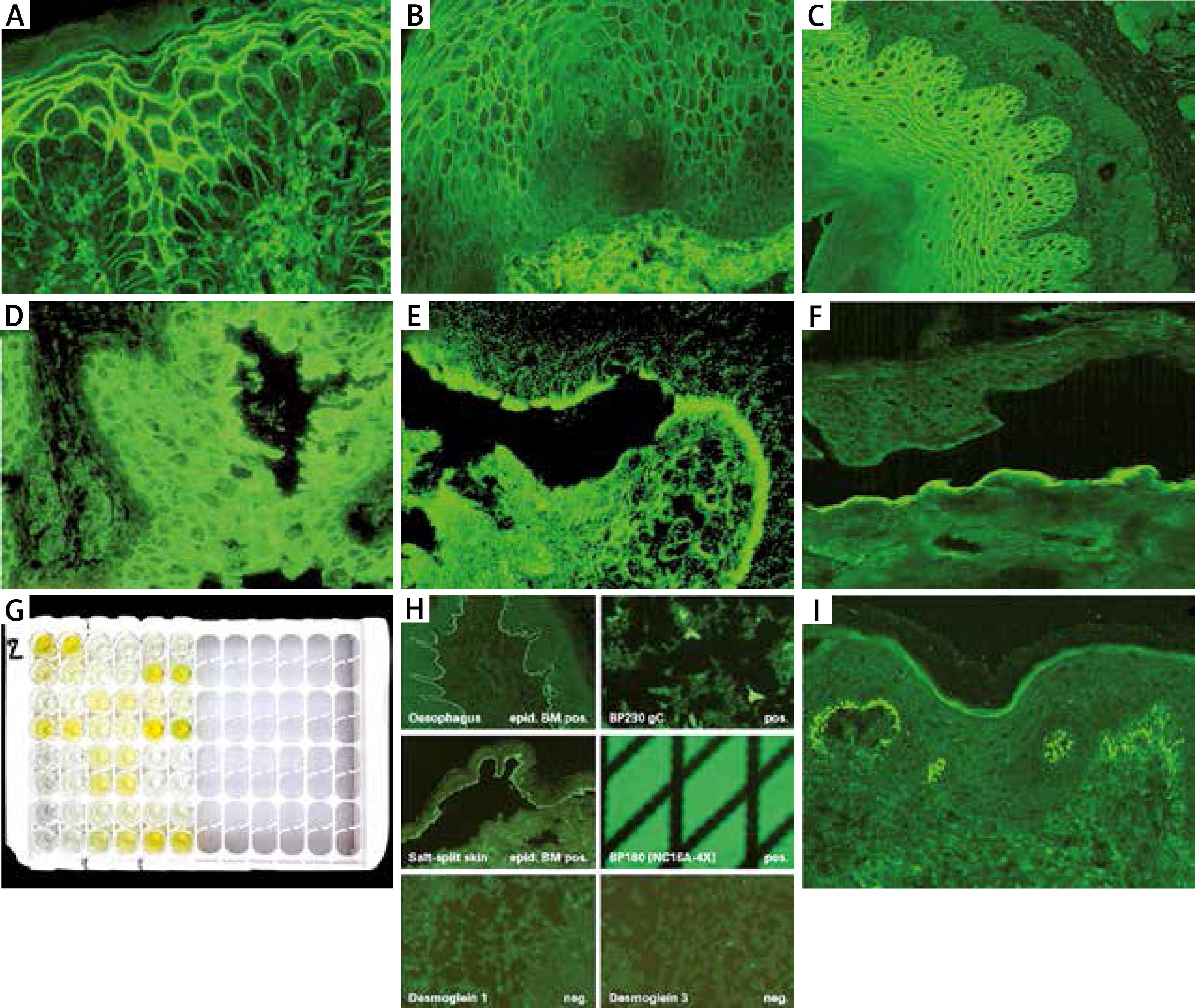
Pemphigus foliaceus
Pemphigus foliaceus (PF) clinically is characterized by the presence of flaccid blisters rapidly transforming into crusted scales. Unlike PV, lesions are present only on the skin, while mucous membranes are free [16]. The diagnostic strategy is the same as for PV. DIF of epidermis reveals the ICS pattern of IgG and sometimes C3. The IIF study reveals fishnet-like pattern of circulating IgG antibodies in ICS on guinea pig oesophagus or human epidermis [17]. Circulating autoantibodies are directed to Dsg1 and can be detected by ELISA or BIOCHIP or immunoblot [12]. Histopathology in PF reveals subcorneal acantholysis, but the test is not obligatory [15].
Paraneoplastic pemphigus
Paraneoplastic pemphigus (PNP) is a rare, life-threatening disorder characterized by the coincidence of neoplasia mostly a lymphoproliferative disease (chronic lymphatic leukaemia, Castleman’s disease). Typical clinical symptoms are painful ulceration in the oral cavity and haemorrhagic crust on the lips, other mucous membranes may also be affected (genital area) [17]. Cutaneous lesions are usually polymorphic and may resemble PV, pemphigoid, lichen planus or graft versus host disease [17]. In some cases, nail changes resembling lichen planus may be present. In contrast to PV, in PNP internal organs like bronchi and urinary bladder are frequently involved still causing PNP life-threatening disorder [17]. DIF in PNP, similarly to PV shows IgG/C3 deposits in ICS or additionally linear IgG deposits along the BMZ. Such a wide clinical symptomatology of PNP is associated with a certain panel of antigens being the aim for circulating antibodies [18, 19]. The most important for the diagnosis of PNP are antibodies directed against envoplakin and periplakin which may be detected by commercially available ELISA or immunoblot [20]. In the pathogenesis of PNP other antigens are also involved, like Dsg3, Dsg1, BP230, desmoplakin 1 and 2, plectin, a2 macroglobulin- like 1 and desmocollins (Dsc) [20]. In many laboratories the IIF test on rat urinary bladder (Figure 1 D) is preferentially used since it is rich in envoplakin and periplakin but not in desmogleins, thus it may differentiate PNP from PV [21, 22]. It is possible to use BIOCHIP mosaic containing a rat bladder as well as monkey oesophagus, Dsg3 and Dsg1, but its validation in PNP patients has not been conducted yet.
Pemphigus vegetans
Pemphigus vegetans (PVeg) is a rare variant of pemphigus vulgaris, characterized by the presence of flaccid blisters which transforms into vegetating plaques. There are two clinical forms of PVeg: Hallopeau and Neumann types. The Hallopeau type demonstrates pustules usually located in axillary folds and groins which heal as vegetative plaques. The Neumann type involves more severe with vegetative lesions of oral mucosa and skin [16, 23]. Immunopathological findings of PVeg are indistinguishable from those of PV although the latest research shows that analysis of Dsc-specific serum IgG and IgA can be valuable in PVeg [24]. In contrast to PV, the histopathology can be helpful in diagnostics of PVeg since when performed from flaccid blister it reveals suprabasal cleft and acantholytic cells similarly to PV, however in vegetating lesions there are observed eosinophilic spongiosis of epidermis, papillomatosis and eosinophilic infiltration of dermis with the lack of intraepidermal blister [23].
Pemphigus mediated by IgA
Thera are two variants of IgA mediated pemphigus (IgAP): subcorneal pustular dermatosis (SPD) and intra-epidermal neutrophilic type (IEN). Clinically, it is characterized by the presence of pustules on erythematous base in SPD, while in IEN pustules form annular pattern [25]. In DIF of perilesional skin, IgA deposits in ICS are present. In SPD autoantibodies are directed against Dsc 1. The antigen of IEN is unknown, although in some cases IgA autoantibodies to Dsg1, Dsg3 or Dsc 1-3 are detectable. IgG and/or C3 deposits can be also present, but they react weaker than IgA. In SPD type reaction between epidermis and IgA it is limited to the upper cell surfaces, whereas in IEN type in lower or entire epidermis. In IIF on monkey oesophagus serum autoantibodies directed to keratinocytes cell surface are present in about 50% of patients. Immunoblotting and ELISA are used to detect serum IgA and IgG against desmosomal proteins such as Dsg or Dsc [26, 27]. In IgAP histopathology shows neutrophilic infiltration, whereas acantholysis is milder than in pemphigus vulgaris or even absent. In SPD subtype, pustules are located subcorneally, while in IEN subtype, intra-epidermal pustules are present (Figures 2 A, B) [25].
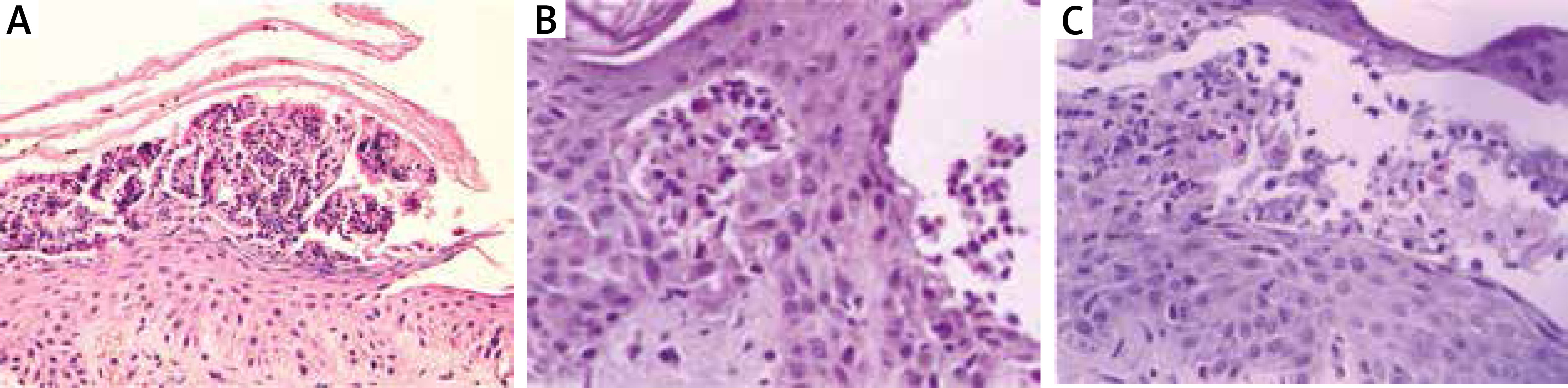
Pemphigus herpetiformis
Pemphigus herpetiformis (PH) combines the clinical features of dermatitis herpetiformis with immunological findings typical for pemphigus. Clinically, PH is characterized by the presence of erythematous, oedematous, vesicular, bullous and/or papular lesions grouped in herpetiform pattern. Mucosal lesions are seen occasionally. DIF shows IgG and/or C3 deposits in ICS like in classic pemphigus. Autoantibodies are directed mainly to Dsg-1, rarely to Dsg-3 and desmocollins and can be detected by IIF, ELISA or immunoblotting. The histopathological findings are very similar to IgA pemphigus with minimal or absent acantholysis and neutrophilic and/or eosinophilic infiltrations (Figure 2 C) [25, 28].
Pemphigoid group
Bullous pemphigoid
Bullous pemphigoid (BP) is the most common AIBD, affecting elderly people over 65 years of age. BP incidence has been steadily increasing in Europe, the USA and Japan as a result of a constant tendency to extend life in the developed countries, progress in medicine and pharmacology [3].
Clinically, BP is characterized by tense blisters, located on unchanged skin or on erythema on the trunk and flexural surfaces of the extremities, they can also be disseminated. Skin lesions are accompanied by severe itching. In about 20% of BP cases the oral cavity is involved [4].
The basis for diagnosis of BP is the presence of linear IgG and C3 deposits along the BMZ in DIF and circulating IgG antibodies along the BMZ in IIF [29]. BP should be differentiated from the inflammatory subtype of epidermolysis bullosa acquisita (so-called pemphigoid-like EBA), disseminated cicatricial pemphigoid, dermatitis herpetiformis and IgA linear bullous dermatosis. To quick and easy distinguish BP from the others one should perform IIF on salt split skin showing the reaction of circulating IgG antibodies with the roof of the artificial blister (Figure 1 E) [30]. For reliable diagnosis it is recommended to demonstrate the autoantibody reaction with the NC16a fragment of BP180 antigen using ELISA or BIOCHIP test, wherein the sensitivity of ELISA is slightly higher than BIOCHIP (82.4% vs. 76.5%) (Figures 1 G, H) [31]. This discrepancy in sensitivity of both methods may be a result of different ways of obtaining the sources of antigens and thus sensitivity of these methods. It is also likely that in some cases, the titre of circulating antibodies is below detectability [32].
Pemphigoid gestationis
Pemphigoid gestationis (PG) usually begins at the end of the 2nd or 3rd trimester of pregnancy. The disease creates the risk of having a baby with blisters on the skin as a result of maternal circulating autoantibodies passing through the placenta to the foetus, which is evidence of their pathogenic role. Circulating antibodies as well as skin lesions in the child disappear within a few weeks. In sporadic cases of PG, premature delivery or miscarriage may develop [33, 34].
The clinical picture of PG consists of urticaria-like lesions and blisters initially located in the umbilical region, then on the buttocks, upper and lower limbs with accompanied itching. Mucous membranes are unoccupied. Skin lesions disappear after delivery but may return in subsequent pregnancies. PG should be differentiated from another disorder of pregnancy named pustular urticarial plaques and papules of pregnancy (PUPPP) [35].
The basis for the diagnosis of PG is finding of C3 component of complement in linear arrangement along the BMZ in DIF. It is also advisable to demonstrate the reaction of circulating antibodies with the BP180 antigen NC16a epitope using ELISA or BIOCHIP. Those tests are negative in PUPPP [36, 37].
Mucous membrane pemphigoid
Mucous membrane pemphigoid (MMP, former name: cicatricial pemphigoid – CP) primarily affects different mucous membranes, mainly of the oral cavity, followed by the conjunctiva. Skin is affected in about 30% of cases. MMP in the oral cavity forms erosions and/or exfoliative gingivitis. The ocular MMP affects approximately 65% of patients. The disease usually begins unilaterally with inflammatory conjunctivitis, a feeling of dryness or the presence of a foreign body, but gradually leads to the involvement of the other eye. Over time, conjunctival scarring occurs, tear duct glandular fouling, and blindness develop in some patients. In 8–20% of MMP cases, mucous membranes of the upper respiratory tract, nose, larynx, oesophagus, genitals and anus are affected causing deformities of the involved tissues. Occupation of the scalp leads to scarring alopecia [38–40].
The basis for the diagnosis of MMP is the finding of characteristic immunological phenomena in the patient’s tissue and serum. In DIF IgG and, in many cases, IgA as well as C3, deposits with a linear arrangement along BMZ are present. For DIF the biopsy should be taken from normal appearing mucous membrane or skin of the affected area. However, if the disease is limited to the conjunctiva the biopsy should be first taken from oral mucosa since in many cases it is positive even if oral mucosa is not involved. Circulating IgG in IIF is detectable in approximately 50% of cases and they are bound to the epidermal site only or both epidermal and dermal sites of artificial blister [41]. Circulating IgG are mainly directed against carboxy-terminal fragment (-COOH) of BP180 antigen which can be demonstrated by immunoblotting in selected laboratories. In some cases, NC16a epitope of BP180 may also be a target antigen demonstrated by ELISA or BIOCHIP [42]. In cases suspected of MMP in which serum tests are negative and it is not possible to characterize the target antigen, it is advisable to use alternative techniques that enable reliable diagnosis, e.g. electron microscopy (which is time consuming) or double immunofluorescence by confocal microscopy (LSCM). Using the LSCM technique, we have clearly shown previously that it was possible to differentiate autoimmune subepidermal blistering disorders on the basis of different localization of IgG/IgA deposits at the BMZ. In contrast to BP (Figure 3 A), MMP is defined by the presence of IgG deposits below laminin 332 and above collagen type IV (Figures 3 B, C). The above-mentioned method should be performed in all cases with erosions located in the oral cavity since they may resemble lichen planus, PV, epidermolysis bullosa acquisita and other inflammatory and allergic disorders [43].
Figure 3
Double immunofluorescence by laser scanning confocal microscopy: A – BP – linear IgG deposits (green colour) localized above laminin-332 (red colour) at the BMZ, B – MMP – linear IgG deposits (green colour) localized below laminin-332 (red colour) at the BMZ, C – MMP – linear IgG deposits (green colour) localized above collagen type IV (red colour) at the BMZ, D – AECP – co-localization of IgG deposits and laminin-332 forming homogeneous( yellow colour) along the BMZ, E – EBA – linear IgG deposits (green colour) localized below collagen type IV (red colour) at the BMZ
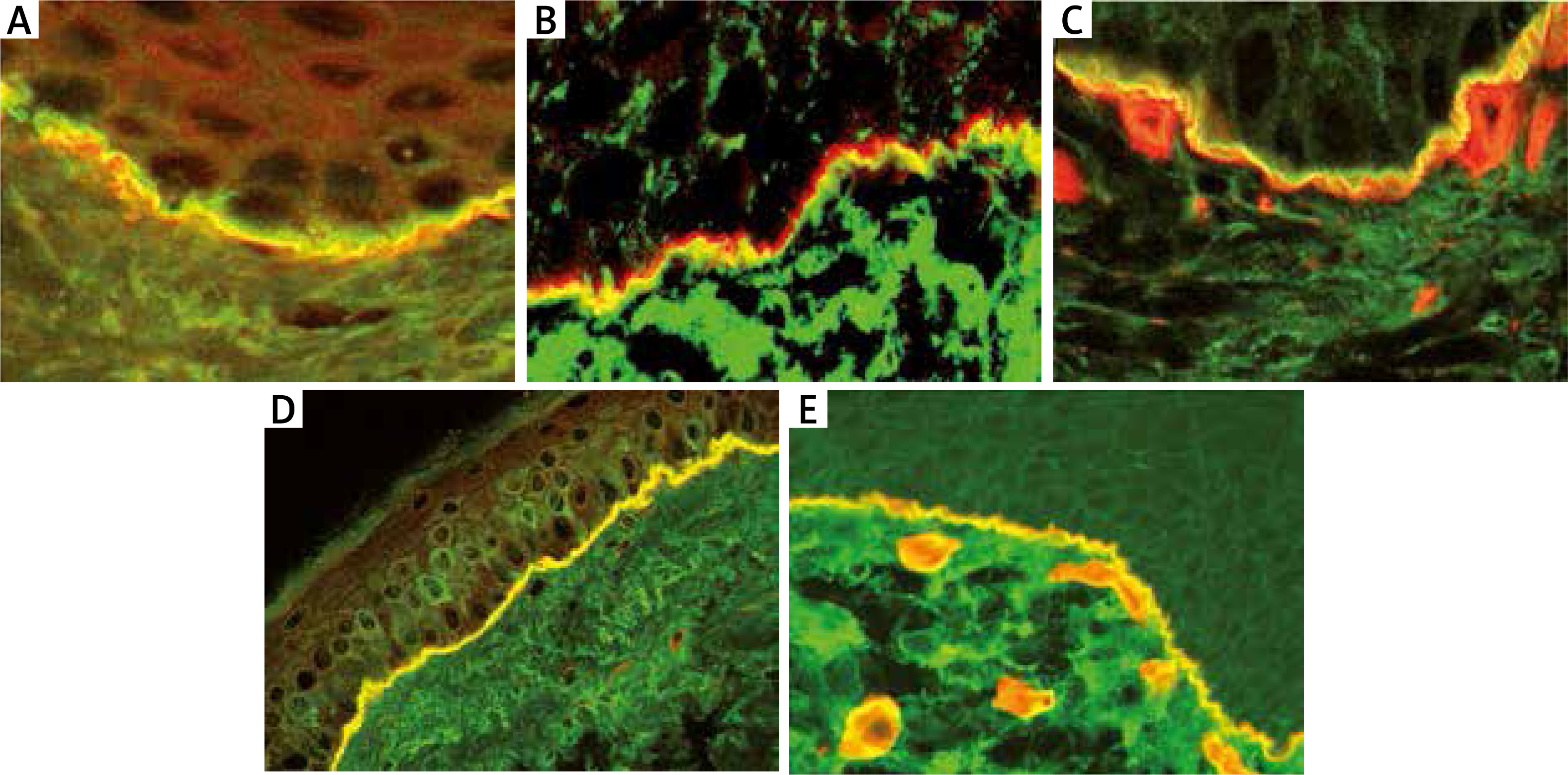
In some cases of MMP (so-called anti-epiligrin cicatricial pemphigoid – AECP), the target antigen is laminin 332 (formerly called laminin 5 or epiligrin). This type of MMP is associated with cancers of internal organs in about 30-40% of cases, therefore a rapid and proper diagnosis is mandatory. The diagnosis of AECP may be established using the IF test with antibodies to laminin 322 (recombinant rabbit polyclonal antibody, Alexa Fluor) or IB (availability limited to a few laboratories) or commercially available ELISA with sensitivity estimated at 75% [44]. In cases with negative sera the diagnosis may be established using LSCM – IgG deposits co-localize with laminin 332 at the BMZ (Figure 3 D) [45].
Epidermolysis bullosa acquisita
Epidermolysis bullosa acquisita (EBA) is a very rare disease in adults. EBA can occur sporadically in children. There are two basic subtypes of EBA: mechanobullous and inflammatory ones. The mechanobullous type is characterized by increased skin fragility and the formation of tense blisters in areas exposed to injuries, mainly on the elbows, knees and backs of the hands and feet. The skin lesions heal leaving atrophic scars and milia. They may also be accompanied by loss of nail plates and narrowing of the oesophagus. In the course of inflammatory EBA, blisters appear on the entire skin often resembling BP [46, 47]. Mucosal involvement may also occur in this variety [48].
Immunologically, EBA is characterized by the presence of antibodies against collagen type VII, responsible for the attachment of the epidermis to the dermis. In DIF IgG and/or IgA as well as C3 deposits are present along the BMZ. In IIF on salt split skin IgG reacts with the bottom of the artificial blister (Figure 1 F) similarly to AECP and anti-p200 pemphigoid [47]. In EBA circulating antibodies react with type VII collagen by ELISA or immunoblot which is detectable in approximately 50% of cases [49]. If serum is negative, it is advisable to use double immunofluorescence by LSCM. In the LSCM technique EBA is clearly defined by the presence of IgG deposits below collagen type IV (Figure 3 E) [50, 51].
There is a subtype of EBA exclusively mediated by IgA (so-called IgA-EBA), which is characterized by the presence of only IgA class immunoglobulins along the BMZ in DIF and IgA circulating antibodies react with collagen VII like IgG antibodies in classic EBA variant [52].
Linear IgA bullous dermatosis
Linear IgA bullous dermatosis (LABD) is a very rare bullous dermatosis, which affects both children and adults. In children LABD appears most often around the age of 5–6 years, while in adults mainly around 50–65 years. In most cases, LABD is idiopathic, but some provoking factors are recognized, i.e. drugs, ultraviolet and irritants [53, 54].
Clinically, LABD in children and adults looks different. In children blisters form so-called “clusters”, most often located on the face around the mouth, the genitals and on the lower extremities. The disease tends to disappear within 2–4 years, therefore it is called self-limiting disease [53]. In adults, skin lesions are more polymorphic and contain disseminated blisters, papules and erythematous patches resembling BP or dermatitis herpetiformis (DH). The oral cavity may be involved [54]. Clinical features are a result of IgA autoantibody reaction with a specific domain of BP180 antigen named LAD-1 [54, 55].
For the diagnosis of LABD, it is necessary to determine in vivo bound IgA along the basement membrane in DIF. The IIF study reveals circulating IgA antibodies, usually at a lower titre than in BP and they react with the roof or artificial blister in IIF on salt split skin and recognize LAD-1 fragment of BP180 antigen by immunoblotting [56]. The histopathological picture of LABD, like the clinical picture, is diverse – in some cases it exhibits DH features, i.e. the presence of neutrophils located mainly in the dermal papillae, in others the presence of sub-epidermal blisters and infiltrates composed of neutrophils and eosinophils along the BMZ, as in BP, and in some patients the histological picture combines the phenomena observed in both BP and DH [52].
Dermatitis herpetiformis
Dermatitis herpetiformis (DH) is a cutaneous manifestation of gluten sensitive enteropathy (GSE). GSE in the course of DH is usually latent and manifests itself after excessive gluten load in the form of diarrhoea. Gluten-dependent enteropathy is more common in children than in adults and it manifests as the disappearance or flattening of intestinal villi [57].
The clinical picture of DH consists of polymorphic skin lesions (papules, vesicles, erythematous, erythematous-oedema lesions, less often blisters) located in typical areas, i.e. scalp, neck, inter-scapular area, elbows, extensor surfaces of the forearm, buttock area, and knees. Skin lesions may be disseminated, reminiscent of eczema or scabies infection and they are always accompanied by severe itching and/or burning of the skin. Mucous membranes are not involved in DH [58, 59].
For the diagnosis of DH, it is crucial to perform the DIF test in a healthy skin biopsy surrounding the lesion and to find the presence of granular IgA deposits in the dermal papillae and/or along the BMZ (Figure 1 I). Some patients with DH have circulating IgA antibodies to smooth muscle endomysium on the monkey oesophagus in IIF, however, this test indicates an intestinal disorder and does not allow to diagnose DH without examining the patient’s tissue. ELISA for tissue transglutaminase (tTG), whose sensitivity and specificity is about 90% in this disease, may be helpful but cannot be the only test in the diagnosis of DH. Histopathological examination of the affected skin shows neutrophil infiltrates in the dermal papillae – this is a characteristic picture of DH, but it is not recommended for diagnosis, because a similar picture can be seen in linear IgA bullous dermatosis [59, 60].







