
The p63 protein is a member of a highly-conserved family of transcription factors (p53/p73) which play a pivotal role in ectodermal and epidermal homeostasis as well as orofacial and limb development [1–4]. Novel data suggest its role in the control of senescence, fertility, hearing, neurodevelopment and even cardiogenesis [5–10]. The gene coding for p63 is located on chromosome 3q28 and consists of 14 exons [11]. It exerts its biologic functions through at least ten different protein isoforms, which are synthesized as a consequence of transcription from alternate promoters (TA and ΔN) and alternative splicing (α-, β-, γ-, δ- and ε-) [12]. The p63 protein is expressed in the nuclei of keratinocytes (predominantly basal ones) of stratified epithelia (skin, tonsils, esophagus and ectocervix), transitional epithelia (urinary tract), simple epithelia (bronchial tree, acini of the breast and prostate, sebaceous and sweat glands), female and male germlines and also to some extent in the germinal centers of lymph nodes [13, 14]. Investigations on p63-deficient animals and humans bearing mutations in p63 shed light on cutaneous morphogenesis and pathophysiology and revealed p63 as a sentinel of epidermal commitment and differentiation, cell-cell adhesion and basement membrane formation [1, 2].
Heterozygous mutations in TP63 cause seven human developmental syndromes, which include: ADULT syndrome (OMIM 103285); ectrodactyly, ectodermal dysplasia with cleft lip/palate syndrome 3 (OMIM 604292); Hay-Wells syndrome (OMIM 106260); limb-mammary syndrome (OMIM 603543); orofacial cleft 8 (OMIM 129400); Rapp-Hodgkin syndrome (OMIM 129400); and split-hand/foot malformation 4 (OMIM 605289), all of which share at least one common feature [15]. However, the influence of specific mutations or positioning of mutated residues in specific domains on TP63-related pathology remains elusive.
Ectrodactyly, ectodermal dysplasia and cleft lip/palate syndrome 3 (EEC3) belongs to group 1 of ectodermal dysplasias according to a novel clinico-molecular classification [16, 17], which not only denotes involvement of two or more ectodermal derivatives but also encompasses its genetic background and pathogenic mechanism (regulatory changes in transcription and/or expression of genes – in this instance p63). We report here a new patient with EEC3 due to a new heterozygous p63 mutation, and discuss the genotypic-phenotypic correlations of this syndrome.
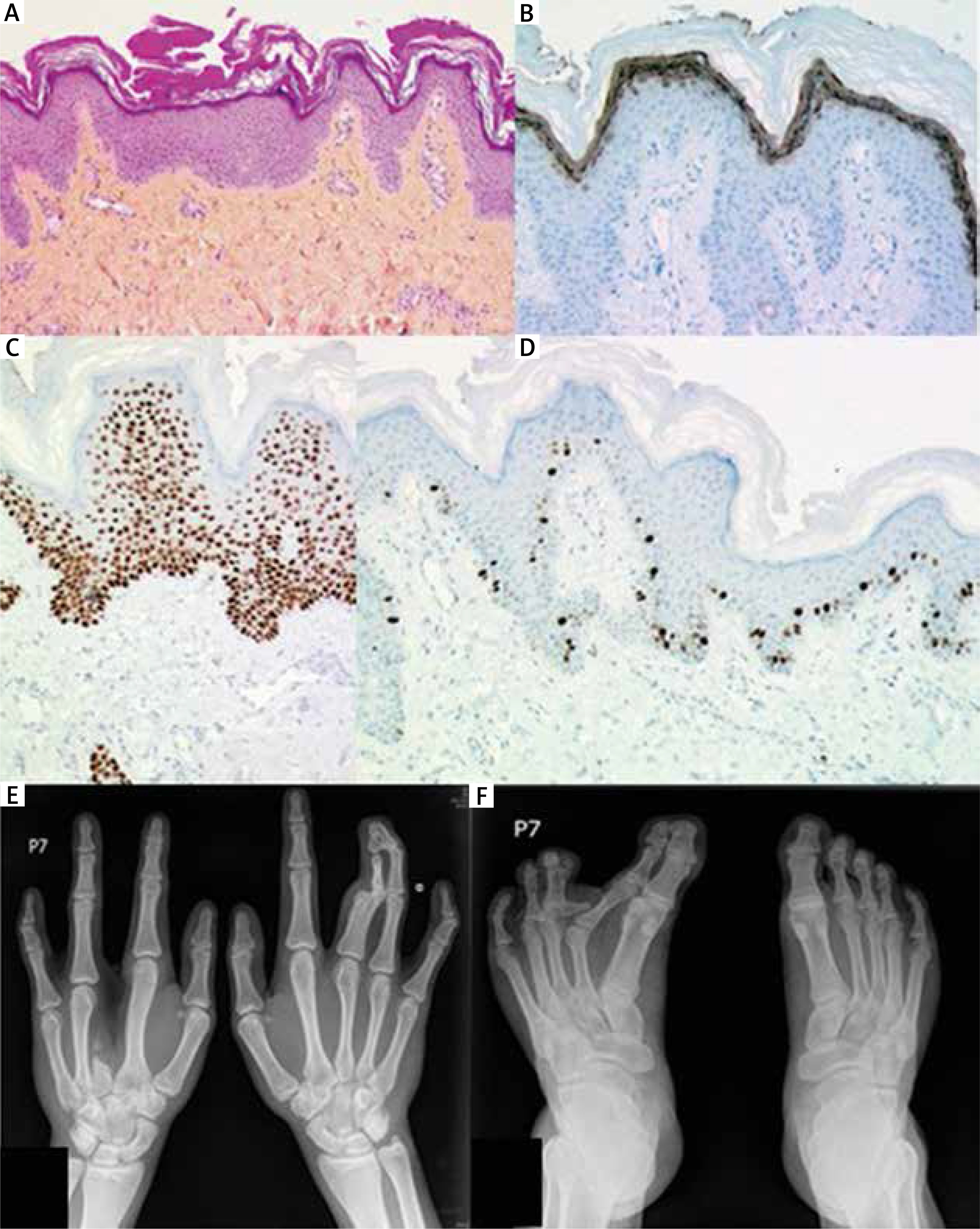
A 23-year-old man was born to unrelated healthy parents with an unremarkable family history for genetic disorders. Physical examination showed sparse, dry, brittle and slowly growing hair (Figure 1). Most of his nails were dystrophic with longitudinal fissuring and ridging, some also with dorsal pterygium. The patient’s skin was fairly pigmented (Fitzpatrick’s type 2) and dry but sweating was reportedly normal. Additional cutaneous findings included psoriasiform plaques over both elbows and widespread (neck, chest, abdomen, elbows, antecubital fossae, wrists, knees, dorsal aspects of the hands and feet) hypopigmented, vitiligo-like macules (Figure 1 G). Histological examination of a psoriasiform cutaneous lesion showed mild psoriasiform epidermal hyperplasia (hyperkeratosis with focal parakeratosis, acanthosis and papillomatosis). The spinous layer contained sparse dyskeratotic keratinocytes (Figure 2 A). PAS staining was negative. Immunohistochemical staining revealed normal expression of p63 in the epidermis and its adnexa (i.e. nuclear labeling of epidermal keratinocytes with a slightly decreasing intensity from the basal to the granular layer, and nuclear labeling of myoepithelial cells and of basal cells of the dermal eccrine sweat-gland ducts) (Figure 2 C). Ki-67 was expressed by some basal epidermal keratinocytes (Figure 2 D). Filaggrin was normally expressed within the granular layer (Figure 2 B). The skin biopsy taken from a hypopigmented macule showed no noticeable changes on routinely stained sections. Immunohistochemical examination showed a normal expression pattern of the p63 protein (as described above). Immunostaining with antibodies to tyrosinase and MART1/Melan-A showed absence of epidermal melanocytes. S100 protein expression was seen in dendritic cells within the suprabasal epidermal layers (Langerhans cells) but not in the basal cell layer (melanocytes) (data not shown).
Figure 1
Trichoscopy shows irregular hair-shaft bending at different angles, as typically seen in pili torti (A), irregularly distributed hair-shaft nodosities (B, red arrows) which appear thicker than the normal hair shaft. Internodes are as thick as a normal hair shaft, as seen in pseudomoniletrix (C, red arrows), longitudinal hair-shaft grooving-pili canaliculi (D, blue arrows), and hair color heterogeneity (A, B, C, D), E – hair of the patient, F – hair shaft microscopic examination with polarized light shows longitudinal grooving and dark and light bands, as seen typically in trichothiodystrophy, G – psoriasiform hyperplasia and hypopigmented lesions of vitiligo on the left elbow, H, I – split hand/foot malformation, J – teeth during treatment
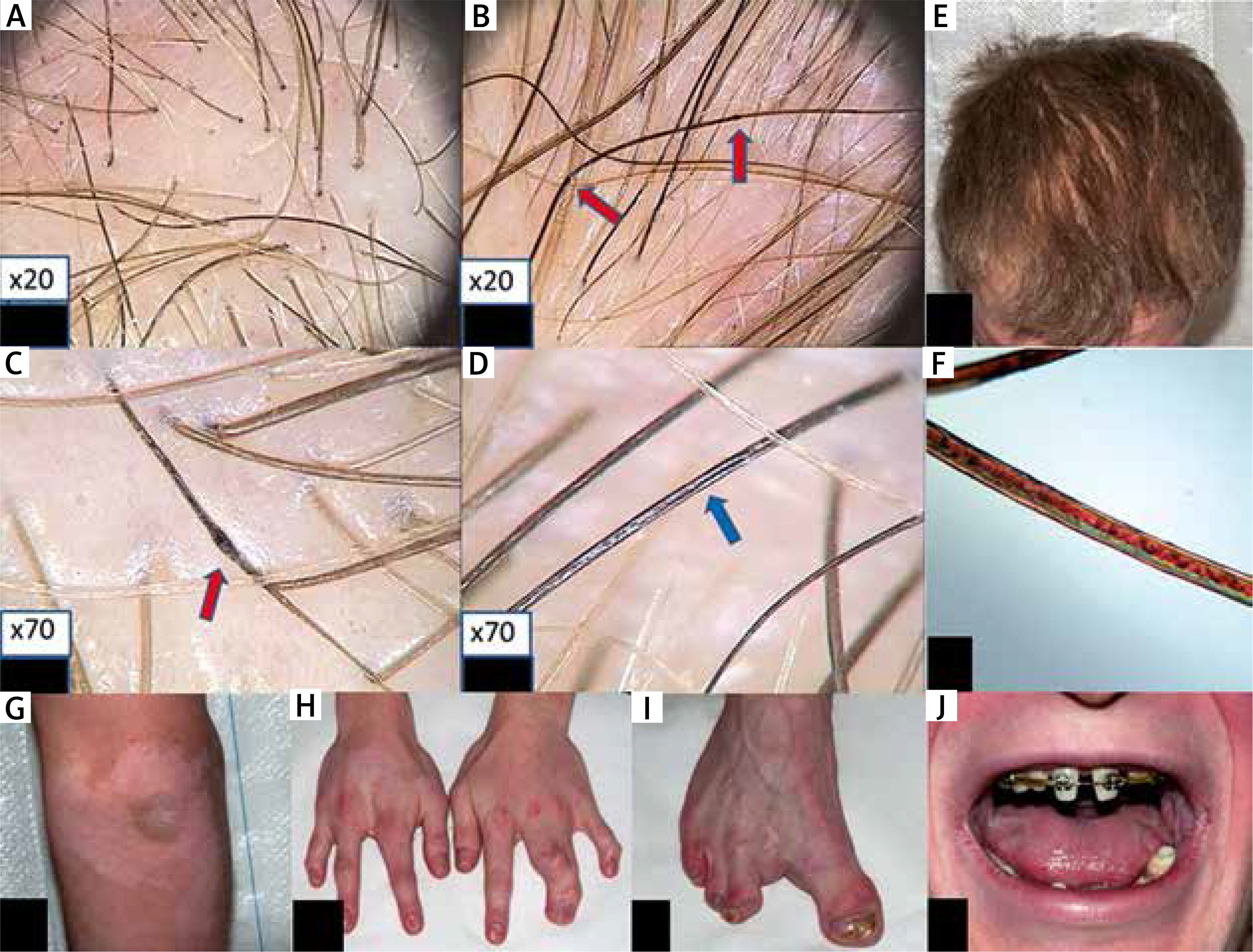
Figure 2
A – Microscopic appearance of a skin biopsy taken from a psoriasiform lesion (hematoxylin-eosin-saffron stain, magnification 100×). B – Filaggrin immunostaining. C – p63 protein expression. D – MIB-1/Ki67 expression (magnification 250×). Radiological examination of hands and feet shows split hand (E) and foot (F) malformation as a consequence of both ectrodactyly and syndactyly
Along with skin findings, the patient presented several extracutaneous manifestations. He suffered since childhood from recurrent kerato-conjunctivitis due to right lacrimal duct atresia and complained of nasal bleeding/crusting and oral dryness. ENT examination and sinus X-rays showed right maxillary sinus hypoplasia. Oral examination revealed retention of deciduous teeth, hypodontia, misshapen permanent teeth but no lip or palate clefting (Figure 1 J). Additional remarkable phenotypic manifestations comprised split-hand and foot deformity of both hands and the right foot as a consequence of syndactyly (left hand and right foot) and ectrodactyly (right hand) (Figures 1 H–I).
Finally, several genitourinary problems impaired the quality of life of the patient; they included recurrent cystitis, hematuria and hypospadias. Abdominal ultrasonography (USG) showed a hypoplastic left kidney and an hyperechogenic, inhomogeneous right kidney. Cystoscopy performed on several occasions revealed hyperemia and exaggerated bladder trabeculae. Renal SPECT tomography and scintigraphy revealed dilatation of the right pyelocalyceal system, interstitial alterations and cortical thinning of the upper segment of the right kidney, moderately impaired glomerular filtration rate, decreased urinary outflow, urinary reflux, urine retention after micturition in the whole kidney (especially in the pyelocalyceal system) and dilatation of the right ureter. Global glomerular filtration rate (GFR) was estimated to 93.1 ml/min – individual values for left and right kidney were 17.9 and 75.2 ml/min respectively.
Skeletal X-rays showed: 1) right hand: ectrodactyly of digit III (lack of phalanges, diaphysis and distal epiphysis of metacarpal bone III); visible epiphysis and small osseous fragment of proximal metaphysis of metacarpal bone III; within soft tissue, two small osseous fragments between metacarpal bones II and IV; 2) left hand: syndactyly (soft tissue) of digits III and IV; deformity of distal part of diaphysis and epiphyseal-metaphyseal segment of proximal phalanx III; synostosis within the proximal interphalangeal joint of digit III; deformity of distal phalanges of digits III and IV; synostosis of distal phalanges of digits III–IV; synostosis within the distal interphalangeal joint of digit IV; 3) right foot: an additional, horizontal arrangement, residual middle phalanx of digit III, connected by additional joint surface to the metatarsophalangeal joint; deformity of the distal phalanges of digits IV and V; syndactyly of digits III–IV and IV–V. Both feet showed shortening of the middle phalanx of digit V with abnormal position of the joint surfaces of the distal interphalangeal joints (Figures 2 E, F).
With informed consent, in accordance with the Declaration of Helsinki and approval from the Medical University Bioethics Committee (approvals no: KE -0254/182/2015 and KE -0254/229/2016), peripheral blood was obtained from the patient and his healthy sister with the purpose of DNA isolation. Molecular analysis of the TP63 gene using direct DNA sequencing showed the missense mutation p.G314E (NP_003713.3: p.Gly314Glu, NM_003722.4: c.941G>A) (Figure 3). This mutation is not yet registered in any database, including HGMD (the Human Gene Mutation Database). It involves a highly conserved nucleotide and amino acid. Bioinformatics analysis using SIFT and MutationTaster software showed the pathogenic nature of the identified mutation. The clinical evaluation of the patient and the results of the genetic testing confirmed the diagnosis of EEC3.
Figure 3
Electropherogram of sequencing results of proband’s DNA (A) and his healthy sister (B). The site of the substitution is indicated by a frame

The EEC syndrome, a member of a larger family of p63-related disorders, comprises three cardinal features: ectrodactyly (absence of the central parts of the hands and feet, also known as split-hand/foot malformation), ectodermal dysplasia, and cleft lip (with or without cleft palate). Not all patients present with the complete triad of these main signs, and the severity of each feature varies among affected individuals [18–24].
Our patient presented the typical constellation of findings of EEC, comprising skin, hair and nail involvement, lacrimal duct obstruction, dental and skeletal malformations, genitourinary malformations and symptoms and oral and nasal dryness, probably due to impaired structure/function of mucous and salivary glands in the setting of ectodermal dysplasia [25].
Hypopigmented and hyperkeratotic-scaly lesions of the skin have been previously reported in EEC patients [26]. In our patient, microscopic examination of the erythematous-scaly lesion showed psoriasiform changes, whereas the hypopigmented macules showed absence of melanocytes, consistent with vitiligo. Despite the fact that a TP63 mutation was present in this patient, the expression of p63 in his skin appeared normal, likely because the epitope recognized by the anti-p63 antibody we used (clone 4A4) was unaffected by the mutation.
The normal Ki-67 and p63 protein expression in the lesional skin of our patient is consistent with previous observations showing that the p63 protein is expressed in the skin despite the presence of a mutation [27–29]. Molecular alterations probably result in reduced transcriptional activity of the impaired p63 on several gene promoters [30–35]. Regarding filaggrin, this was normally expressed in the skin of our patient. Candi et al. [36] found that p63–/– animals had decreased, but still measurable, filaggrin (along with K14 and loricrin) expression, suggesting that their production is regulated by genes other than TP63.
Although genitourinary abnormalities are not common (or often overlooked) in EEC, they may cause non-negligible morbidity. The most commonly reported abnormalities include absent or hypoplastic kidneys, hydronephrosis, hydro-ureter, duplication of the urinary collection system, hypospadias and cryptorchidism [37–40]. Many patients complain of micturition problems and recurrent cystitis. Bladder biopsies show thin, atrophic or dysplastic bladder epithelium, or interstitial cystitis [39, 40]. Our patient had both structural and functional disturbances, possibly reflecting interrupted epithelial-mesenchymal interactions by the mutant p63 [41–43].
Analysis of genotype-phenotype correlation in 152 patients with p63-associated EEC revealed hair, teeth, nail and lacrimal duct involvement in about half of them [19]. Cleft lip/palate was present in 39% and 40% of cases respectively, ectrodactyly in 68% (43% also had syndactyly), skin involvement in 34%, hypohidrosis in 11% and urinary tract problems in 15%. Mammary gland/nipple hypoplasia and hearing impairment were found in 11% and 7% of patients, respectively. The variable penetrance (the average incidence of any cardinal sign does not exceed 50%) seen in EEC syndrome can be partially explained by the specific effects of different mutations. Five arginine hotspot mutations (according to the novel HGVS nomenclature: p.Arg243Trp, p.Arg266Gln, p.Arg318His/Cys/Ser, p.Arg319His/Cys/Ser, p.Arg343Trp/Pro) located in the DNA-binding domain (DBD) region of the p63 protein account for almost 90% of all known mutations [21]. For instance, patients bearing the p.Arg243Trp mutation usually present with typical EEC findings, but have a lower incidence of orofacial clefting (26% vs. 40%) and hypohidrosis; those with the p.Arg227Gln mutation are less frequently affected by orofacial clefting (cleft lip 0%, cleft palate 7%) and limb defects (40% vs. 68%), but have a higher risk of genitourinary complications (39%). Conversely, the p.Arg304Trp/Pro mutation is associated with orofacial clefting in 80% of cases, whereas this reaches on average 40%.
Our patient had a new p.Gly314Glu heterozygous mutation of the TP63 gene, yet with signs and symptoms comparable to the typical clinical presentation, with the closest clinical similarities to individuals with p.Arg243Trp R204 and p.Arg266Gln R227 mutations.
We can also presume that cutaneous, craniofacial, long-bone and genitourinary symptoms depend partly on p63-response elements. BMP7, FGFR2B, JAG1, NOTCH1, B-catenin, EDAR, P-cadherin, DLX5, DLX6, and IRF6 are only some of up to 328 genes whose expression is altered in mutant EEC keratinocytes [2, 33–35]. It was shown recently that the two transcription factors p63 and BRG1 form a complex network of reciprocal interactions controlling remodeling of the higher-order chromatin structure of the epidermal differentiation complex locus [44] and the urothelial cell fate during ureter development [42].
Establishing strict genotype-phenotype correlations is complicated for several reasons, including varying expression and ability of each p63 isoform to transactivate or repress p63-dependent genes, genetic background, epigenetic modifications, the impact of mutant/wild type ratio on organ involvement and subtle genetic alterations with a striking phenotype shift from one side and the same mutation producing distinct manifestations on the other [31–33, 36, 37]. For instance, recent research on genetically engineered mouse models of EEC syndrome found that animals heterozygous for Trp63 encoding the same single amino acid mutation, which differed only in the presence or absence of the neomycin cassette within intron 4, displayed marked heterogeneity regarding development, penetrance and symptom severity [32]. Furthermore, based on the same allelic series of mouse EEC model, it was found that (contrasting with clefting and skin pathology, which reflected loss of Trp63 function), limb anomalies were due to gain and/or dominant-negative effects of Trp63. Moreover the TA-p63 isoform was found to be an important factor modulating EEC phenotype penetrance and expressivity regarding all three cardinal EEC features [32].
In conclusion, the molecular basis of EEC seems complex. Our patient carried a new p.Gly314Glu heterozygous mutation of the p63 gene that affects the sequence of exon 7, which encodes the DBD (amino acid residues 123–340 of the p63 protein) [45]. The glycine at position 314 is conserved in two other TP53 gene family members, i.e. TP53 and TP73 (at positions 243 and 264, respectively). It does not directly contact DNA; however, prediction of DNA-protein interaction in the p63 protein model proposed by Celli et al. [46] has shown that mutations within this region might alter the positioning of the p63-binding surface; this in turn may result in diminishing the DNA-binding capacity of p63 and abolishing its activity as a transcription factor [46, 47].
The location of the mutation site influences the clinical presentation of the disease. Studies of genotype/phenotype correlations in EEC are additionally hindered by its reduced penetration and variable expressivity, which accounts for phenotypic variability in the affected members of the same family. The identification of the novel p.Gly314Glu mutation further expands the knowledge concerning the causes of EEC, and confirms the necessity of sequencing the TP63 gene (primarily exons 4–8) in patients presenting with symptoms, in order to identify the leading cause.







