
Introduction
Psoriasis is a chronic inflammatory disease with gene predisposition, which can be triggered by environmental factors. Pathogenesis is characterized by an activation of Th1/Th2 axis and abnormalities of the Th17/Treg balance [1]. An essential factor of pathogenesis is the dysfunction of regulatory lymphocytes (Tregs), which are involved in homeostasis mechanisms to maintain tolerance and prevent autoimmune disorders [2].
Tregs are a heterogenous subpopulation of lymphocytes responsible for suppressing an autoreactive immune response (Table 1). They interact directly through membrane receptors for immune cells (T-lymphocytes, memory and NK, B lymphocytes, antigen presenting cells), by secretion of suppressing cytokines (IL-10, IL-35, TGF-β, galectin-1) or by direct cytotoxic action (granzyme B and perforin release) [3–6]. Phenotypically, Tregs express high levels of FOXP3, cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), certain members of toll-like receptors, CD103 and glucocorticoid-induced TNF receptor family-related gene (GITR) [7–9]. FOXP3 is a more specific marker of Treg, which induces GITR, CD103 and CTLA-4 [9, 10]. Lymphocyte Tr1 cells depend on IL-10 for their induction and suppressive action, whereas Th3 cells depend on TGF-β for suppressive action. Some Tregs can be induced by IL-35 (iTr35) [10, 11] (Table 1).
Table 1
The characteristics of CD4+ Tregs subtypes
Some researchers found deviations in Tregs in peripheral blood of psoriatic patients, which led to hyperactivity of T effectors in vivo [12]. After an effective biologic therapy, an increase in Tregs in blood of psoriatic patients is observed [13, 14]. However, the literature shows a lot of contradictory reports. Some authors did not find any differences in the percentage of Tregs between psoriatic and healthy patients [12, 15, 16].
The discrepancies of the above studies indicate that not the number of Tregs, but their dysfunction may be significant in psoriasis pathogenesis. Tregs isolated from the psoriatic lesions or peripheral blood are functionally deficient in suppressing T effectors cells and are not capable to suppress Th1 in psoriatic patients [17, 18]. In contrast, those isolated from peripheral blood of healthy controls are able to inhibit psoriatic Th1 in vitro [3, 7, 19]. Thus, the dysfunctional Treg cell activity in the blood and psoriatic plaques may eventually result in the reduced restraint and hyperproliferation of psoriatic pathogenic cells in vivo [17, 18].
Interleukin-10 has anti-inflammatory properties, inhibiting the production of pro-inflammatory cytokines (IFN-γ, IL-2, IL-3, TNF-α, GM-CSF). It is produced by Tregs, macrophages, dendritic cells and B lymphocytes. Interleukin-10 inhibits the production of IL-12 by macrophages, which is responsible for Th1 maturation and IFN-γsecretion [20]. In psoriasis, a relative deficiency of IL-10 in serum and skin is observed, therefore it seems to be an important factor in psoriatic pathogenesis [21].
Transforming growth factor-β is secreted by Treg populations, especially Th3, being an important regulator in immune homeostasis. Moreover, it can limit keratinocyte hyperproliferation in psoriasis [22, 23].
Several studies confirmed an increased TGF-β1 expression in lesions and serum in psoriatic patients. TGF-β1 levels correlated with the severity of the disease [22]. However, based on clinical data, it is difficult to determine whether increased TGF-β1 has a causative function in psoriasis or is a result of the inflammation development. Contrary to plasma levels, the results of TGF-β1 in psoriatic lesions are still contradictory.
Another crucial part of analysis on the role of TGF in psoriasis is assessing its isoforms (TGF-β1, TGF-β2 and TGF-β3), mediated by specific receptors (TGFβRI, TGFβRII and TGFβRIII) [24]. Since TGF-β is a potent growth inhibitor for human keratinocytes, the decrease of TGF-β2 in the epidermis of psoriatic skin may contribute to epidermal hyperplasia. Moreover, it is confirmed that TGF-β2, and not only TGF-β1, induces FOXP3 expression in CD4+CD25+ precursors. Therefore, two isoforms of TGF-βhave been reported to have similar biological effects [24, 25].Considering much lower binding affinity of TGF-β2 to TβR-II than TGF-β1, TGF-92 may be relatively more potent in inducing FOXP3 expression [25]. Cai et al. revealed that both TGF-β1 and TGF-β2, and, to a lesser extent, TGF-β3 isoforms block the ability of normal but not psoriatic dermal microvascular endothelial cells to bind lymphocytes, as the first stage of inflammation cells migration psoriatic lesions [26]. Therefore, we decided to assess TGF-β2 in our study.
Interestingly, the role of IL-35, which belongs to the IL-12 family, has not yet been studied in psoriasis. It is produced mainly by Tregs and downregulates Th17 development, suppressing IL-17 production and promotes IL-10 expression [4, 27–30]. Moreover, some studies suggest that it may reduce the proliferation of CD4+ cells [27]. Zhang et al. found that IL-35 decreased the number of macrophages, and compared with dexamethasone, IL-35 showed long-term therapeutic efficacy. Thus, it can be a new therapeutic strategy for psoriasis and other cutaneous inflammatory diseases [30]. However, the role of IL-35 in psoriasis remains unclear.
Aim
In this study we evaluated the expression of protective cytokines in serum and Treg markers in lesions and perilesional skin in men with psoriasis in comparison to healthy volunteers.
Material and methods
Study group
The study group consisted of 33 Caucasian males with severe plaque psoriasis, without psoriatic arthritis (Table 2). They were recruited according to the Declaration of Helsinki principles. Patients with chronic and acute inflammatory diseases, cancer, cardiovascular diseases, renal and hepatic failure (n = 3) have been excluded. Patients were not treated: a wash-out period for topical treatment was 2 weeks, 4 weeks for photo(chemo)therapy and systemic therapy, biologics were not used at all. The control group consisted of 6 males, healthy volunteers without personal or family history of psoriasis. We decided to choose only males, because oestrogen levels can change Treg function and alter the cytokines profile [31].
Table 2
Patient demographics
| Parameter | Psoriatic | Healthy controls |
|---|---|---|
| N | 33 | 6 |
| Age [years] | 46.45 (18–66) | 47.3 (21–74) |
| PASI | 19.58 (10.2–45.6) | – |
The permission of the Bioethical Committee of the Warmia and Mazury University was obtained (33/2015).
Tissue specimens
We obtained two 3-mm punch biopsies per patient: one from the centre of the psoriatic plaque (abdominal region), one from non-lesional skin (at least 2 cm from the target lesion), and one from healthy volunteers (abdominal region), using local anaesthesia (2% lignocaine). We assessed 66 biopsies from psoriatic patients and 6 from controls.
Enzyme-linked immunosorbent assay (ELISA)
Determination of cytokines (IL-35, IL-10, TGF-β) was performed using the available kits – for IL-35 with the detection range of 15.6–1000 pg/ml (Wuhan Fine Biological Technology, China), for IL-10: 1.56-50 pg/ml (Diaclone, France) and for TGF-β1: 1.2–600 pg/ml (Demeditec Diagnostic GmbH, Germany), according to the manufacturer’s protocol. Validity of the assays was confirmed by parallelism between the standard curves and series of dilutions of randomly chosen plasma samples. The intra-assay coefficient of variation was < 5%. The inter-assay factor of variations was not calculated (all analyses were done in one assay). The sensitivities of the assay were 9.4, 0.98 and 1.9 pg/ml for IL-35, IL-10 and TGF-β1, respectively. We assessed sera from 31 psoriatic patients and 6 from controls.
Immunohistochemistry
The skin biopsies were dissected into 4-μm thick sections using cryostat CM3050 (Leica, USA) and mounted onto poly-L-lysine-coated glass microscope slides (Menzel-Glaser, Braunschweig, Germany). Frozen sections after reaching room temperature were rinsed three times in 0.01 M PBS and incubated with 3% H2O2 in methanol for 30 min. Next, to decrease nonspecific binding, they were incubated with 5% bovine albumin serum (Sigma Aldrich, USA) for 60 min at room temperature. The biopsies were incubated with rabbit polyclonal antibody to IL-27/IL-35, FOXP3, TGF-β2 and IL-10 (1 : 50; NovusBio, USA) and mouse monoclonal antibody to CD4, CTLA-4, CD25/IL-2R, CD39/ENTPD1, IL-7R/CD127 and CD3 (1 : 50; NovusBio, USA) diluted in PBS at 4°C overnight. The following day, the biopsies were washed three times in PBS and incubated with secondary anti-mouse or anti-rabbit antibodies (1 : 200; Jackson ImmunoResearch, USA) for 60 min. To visualize the immunoreactions, they were immersed in 3.3′-diaminobenzidine (DAB, Dako, USA), next they were dehydrated in ethanol, cleared in xylene and mounted with DPX (Sigma Aldrich, USA). The labelled tissues were photographed using a C-5060 Camera (Olympus, Japan) mounted on a light microscope (CH30/CH40, Olympus, Japan).
The images were subjected to semiquantitative analysis. Since the parameter defying staining intensity did not differ between the examined stages, the assessed parameter was the immunoreactive area (%). It was calculated as the ratio of area occupied by the immunopositive cells to the total area occupied by tissue. The level of immunoreactivity was measured with ImageJ software (image processing and analysing in Java, USA). Quantitative analysis was performed using an automatic threshold function to select a range of grey values that were optically identified as positive staining. Before statistical analysis, percentage data were arcsine transformed.
Results
Concentration of IL-10 and IL-35 in serum are not statistically different in patients with psoriasis and the control group
The serum levels of IL-35 (Figure 1 A) and IL-10 (Figure 1 B) were higher in psoriatic patients, but without statistical significance. Interleukin-10 levels were determined in the range of 1.5 ±0.32 to 4.4 ±1.45 pg/ml (p = 0.56) and IL-35 of 13.7 ±0.84 to 17.0 ±1.38 pg/ml (p = 0.40), respectively. There was no statistically significant relationship between PASI and IL-10 (r = 0.0115), IL-35 (r = –0.0011). Correlation analysis showed a statistically significant relationship between IL-10 and IL-35 levels (r = 0.8427).
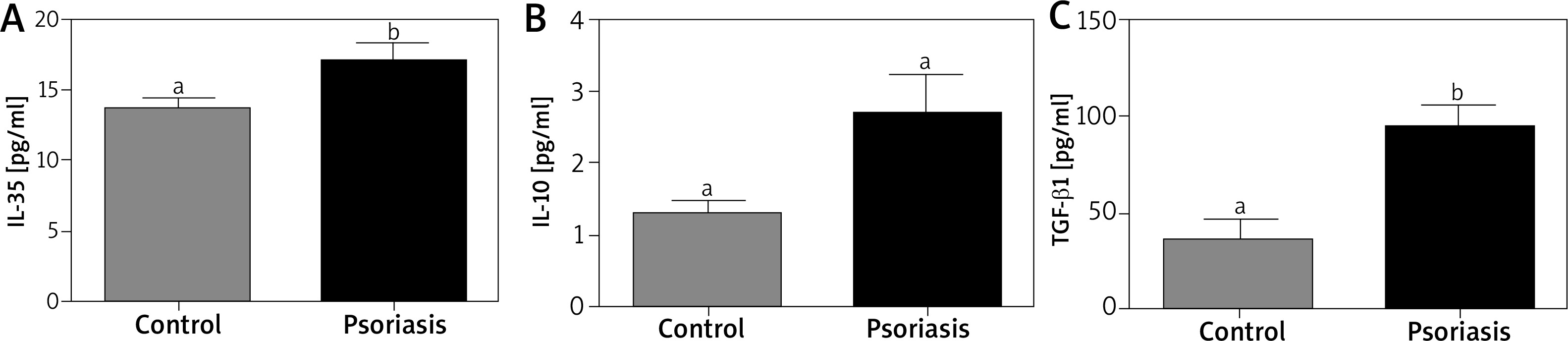
Concentration of serum TGF-β1 is increased in the serum of patients with psoriasis
Concentration of TGF-β1 revealed variations (Figure 1 C) and a higher (p < 0.05) level of TGF-β1 was noted in the psoriatic patients than in controls (96.1 ±8.62 pg/ml vs. 34.9 ±3.96 pg/ml). There were no statistical correlations between PASI and TGF-βlevels (r = 0.0156).
Immunoreactivity for IL-35, IL-10, TGF-β2, CD4, CD3, FOXP3, CD25/IL-2R, CTLA-4, IL-7R/CD127 and CD39/ENTPD1
The expression of all analysed proteins was noted in three experimental groups (lesional, perilesional and healthy skin). The level of immunoreactive IL-35, IL-10, TGF-β2, CD4, CD3, FOXP3 and CD25/IL-2R proteins varied in different experimental groups (p < 0.05). The level of IL-35 was the lowest in the skin biopsies from psoriatic lesions (p < 0.05) compared to healthy and perilesional skin (Figure 2 A). The level of CD4 (Figure 2 B), IL-10 (Figure 2 C) and TGF-β2 (Figure 2 D) were higher (p < 0.05) in perilesional skin than in lesions. TGF-β2 expression was decreased in lesional and perilesional skin compared to controls (Figure 2 D, p < 0.05).
Figure 2
The immunoreactive area of IL-27/IL-35 (A), CD4 (B), IL-10 (C), TGF-β2 (D), CD3 (E), FOXP3 (F), CD25/IL-2R (G), CTLA-4 (H), IL-7R/CD127 (I) and CD39/ENTPD1 (J) proteins in the sections from healthy skin (n = 8), perilesional skin (n = 33) and skin with psoriatic lesions (n = 33). Bars with different letters are significantly different (p < 0.05). The proteins are marked in brown (3,3’-diaminobenzidine – DAB). Nuclei were stained with hematoxylin (violet) (magnification 400×)
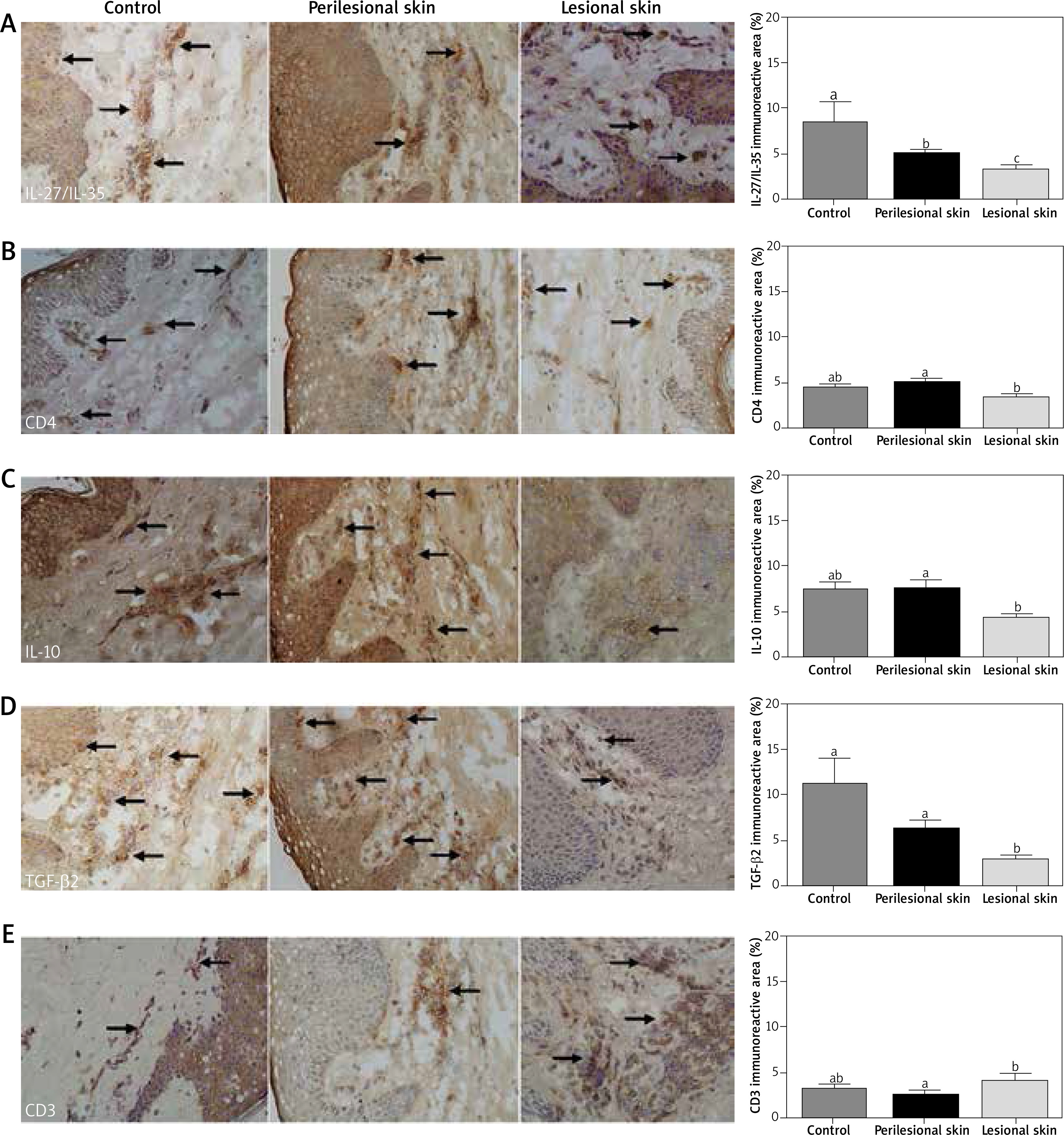
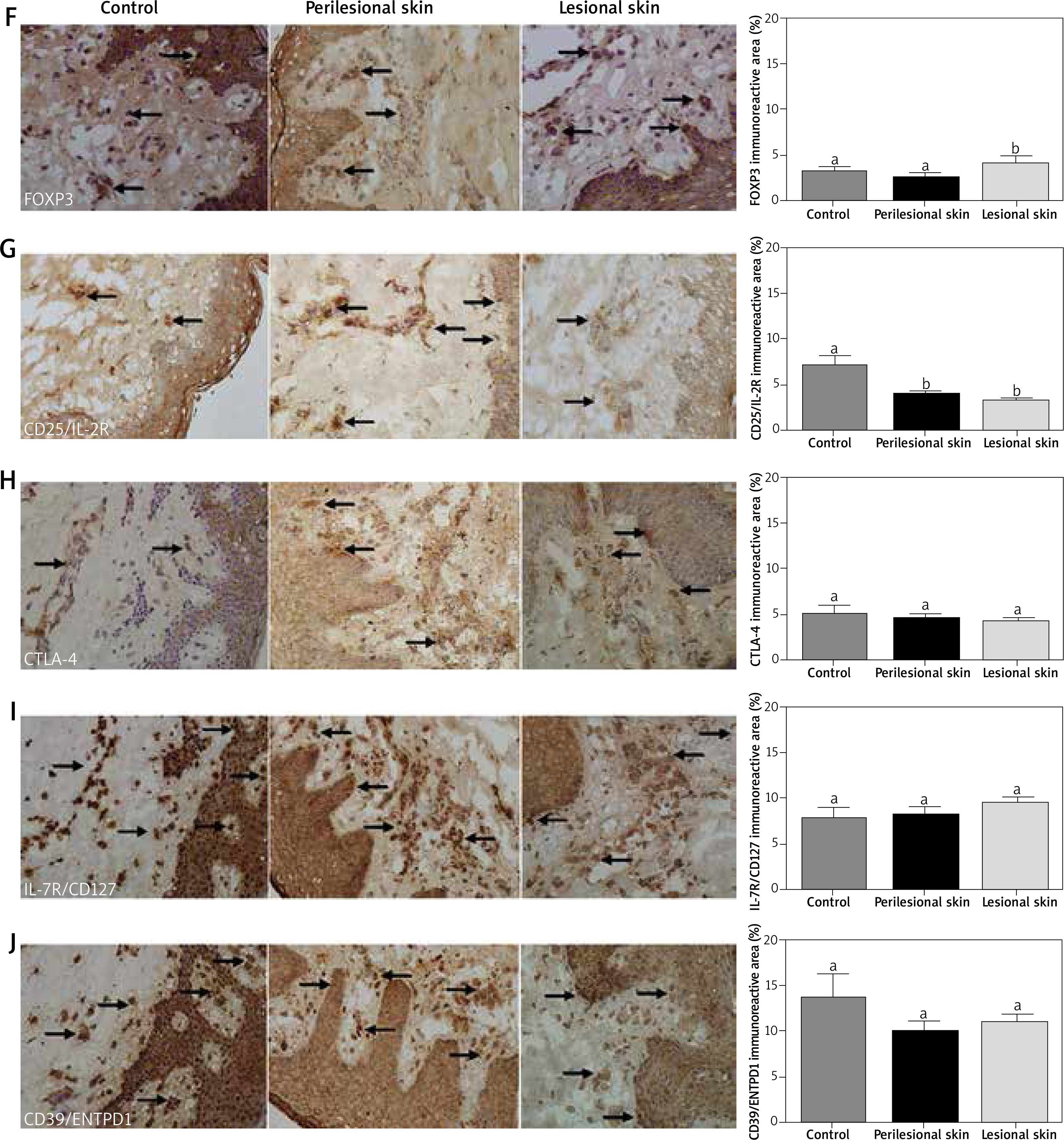
The variance of CD25/IL2R (Figure 2 G) was characterized by the increase in healthy skin (p < 0.05) compared to perilesional and lesional (p < 0.05) (Figure 2 G) with no differences between them. On the contrary, the elevated level of FOXP3 expression was noted in lesions only (Figure 2 F). There was no difference between experimental groups in CTLA-4 (Figure 2 H), IL7R/CD127 (Figure 2 I) and CD39/ENTPD1 (Figure 2 J) protein expression.
There was a close correlation between IL-10 and IL-35 both in the controls (r = 0.9099), perilesional (r = 0.5236) and lesional skin (r = 0.4514). TGF-β2 expression was higher (p < 0.05) in the perilesional epidermal and dermal layer than in psoriatic lesions (Figure 2 D).
Expressions of CD25, CD3, FoxP3, CD39, IL-10, IL35 and CTLA-4 were higher (p < 0.05) in the dermis, both in the papillary and reticular layer. There were no significant differences in the expression of TGF-β2 and IL-7R between skin and epidermis (Table 3).
Table 3
Expression of Treg markers and protective cytokines in the healthy, lesional and perilesional psoriatic skin – immunohistochemistry
Discussion
We observed that the levels of IL-10 and IL-35 in serum are higher, but not statistically different from patients with psoriasis and the control group. Similarly to our results, Borska et al. reported that IL-10 levels were significantly higher in psoriatic patients, and decreased after the treatment [32]. The same results were obtained by Deeva et al. without any correlation between PASI and IL-10 in mild-to-moderate psoriatic patients [33].
In contrast to our results, other authors found a significant decrease in serum IL-10 levels in psoriatic patients compared to healthy controls [34, 35]. Verghese et al. found that IL-10 levels were higher in controls than in patients without statistical significance [36]. Similarly, Takahashi et al. detected decreased serum levels of IL-10 in psoriasis which negatively correlated with PASI [37]. However meta-analysis of 6 researches showed a small, positive but not statistically significant difference between psoriatic patients and controls. It did not depend on age, sex, PASI or psoriasis type [38]. These controversial results do not confirm IL-10 as a reliable biomarker for psoriasis.
Despite the significant anti-inflammatory effect of IL-10 and reports of its effectiveness in psoriasis therapy, the results of serum psoriasis studies are not conclusive. However, the genetic polymorphism of IL-10 may affect the expression of this cytokine.
In the literature, there are a few reports about IL-35 in psoriasis. Cardoso et al. did not find any differences between the level of IL-35 in the serum of Brazilian psoriatic patients and healthy controls [27]. Li et al. also showed that serum levels of IL-35 were higher in patients with psoriatic arthritis (especially in erosive phases) just as in healthy controls. This result suggested that IL-35 was closely related to arthritis [29].
We assessed significantly higher levels of serum TGF-β1 of patients with psoriasis than controls as some other researchers [22, 39–42]. Kallimanis et al. observed a decrease in TGF-β1 together with a decrease in PASI after biological treatment. Hence, TGF-β1 levels seem to be sensitive to changes in disease severity [39].
On the other hand, Cardoso et al. did not find statistical significances for TGF-β in serum of Brazilian psoriasis patients and healthy participants [27]. Similarly, Zaher et al. observed that the mean serum levels of TGF-β1 in psoriatic patients were higher than controls but statistically non-significant, although correlating with PASI [42].
Probably the increased TGF-β1 could be a result of activated endothelial cells, fibroblasts, or inflammatory cells which can produce TGF-β1 in psoriasis [22].
Our immunohistochemical investigations suggested a low cutaneous IL-10 expression. Similar results were found by quantification of IL-10 protein in blister fluids [21, 43, 44]. However, conflicting data have been published. Overexpression has been reported by some authors, but the groups of patients were too small [45, 46].
There is no study evaluating IL-35 expression in psoriasis. We found a close correlation between IL-10 and IL-35 both in the controls, perilesional and lesional skin. It suggested an anti-inflammatory role of IL-35 in psoriasis.
We found a higher TGF-β2 expression in the perilesional epidermal and dermal layer than in psoriatic lesions. Wataya-Kaneda et al. found a decreased amount of TGF-β2 in the psoriatic epidermis, which contributes to epidermal hyperplasia. The intensity of immunoreactivity has the tendency to decrease in the lower epidermis rather than in the upper epidermis of the transitional lesion [47]. Flisiak et al. revealed no correlation between PASI and TGF-β1, TGF-β2 in psoriatic lesions with a significant correlation only between TGF-β1 concentration in scales/size and the disease duration [48]. The study of Yu et al. demonstrated a significant activity of TGF-β3 and TGFβRII mRNA only in non-lesional psoriatic skin but there was no significant difference in the expression of TGF-β1 and TGF-β2 [49].
Interestingly, our results revealed a reduced expression of IL-10, IL-35 and TGF-β1 in psoriatic lesions compared to perilesional skin. This may indicate their involvement in inhibiting inflammation in psoriasis and their amount decreases due to their wear off. It suggests an anti-inflammatory role of IL-35 in psoriasis, although it belongs to the proinflammatory group of the IL-12 family.
We confirmed a higher concentration of CD3 (co-receptor of cytotoxic T-cells and Th) in psoriatic lesions compared to perilesional skin. Similar results were obtained by other authors [50–52]. This is a typical phenomenon associated with the involvement in psoriatic inflammation of Th1, Th17 and Th22. Interestingly, in our study, the concentrations of CD4 proteins were higher in perilesional skin than in lesions.
To evaluate Treg activity, an assessment of CD25 and FOXP3 expression was made. However, the intracellular location of FOXP3 limits its usefulness in studying and purifying this subpopulation. Cell surface CD25 expression is often used as a marker for CD4+Tregs, but CD25 is somewhat nonspecific [53]. There are only a few studies devoted to the evaluation of other markers of Treg in the literature, including CTLA-4, chemokine receptors (CCR4 and CCR8), selectins (CD62L), integrins (CD103), and CD127. In vivo, putative mechanisms include the release of soluble factors IL-10 and TGF-β [53, 54]. Their role is not fully understood in psoriasis. We made an assessment of some of the most important ones, bearing in mind the heterogeneity of the Treg group.
Our results were similar to the other studies. Psoriatic lesions exhibit an increased FOXP3+ expression compared to the healthy skin [18, 19, 55, 56]. There are an increased number of FOXP3+Treg in psoriatic lesions compared to controls and they were located predominantly in the papillary and upper reticular layers of the dermis [56]. We observed an elevated expression of FOXP3 in lesional dermis compared to perilesional skin. However, in psoriatic patients, Tregs readily turn into IL-17-expressing cells, which potentially perpetuate the inflammatory process that characterizes the disease [57]. Bovenschen et al. proved that psoriatic Tregs exhibit a propensity to alter levels of the main regulator FOXP3 and upregulating the expression of RORγt by the addition of proinflammatory cytokines characteristic for psoriatic lesions (IL-2, IL-15, IL-1β, and IL-23), together with a decrease in FOXP3 and an increase in IL-17-producing Treg. Moreover, the conversion from Treg to IL-17/Treg is a continuum of converting cells which exists, as evidenced by FOXP3+ RORγt+ co-expression and a gradual loss of FOXP3 [19]. In this way we can explain the lack of differences in the expression of other Treg-specific molecules in the study groups. What is more, the expression of CD3, characteristic of T-cell glycoprotein, has been found higher in the dermis.
However Keijsers et al. found a significant increase in CD3+, CD4+ and FOXP3+cells in the transition from the symptomless to lesional skin. They were higher in the distant uninvolved skin than in the perilesional, lesional skin and even higher than in healthy skin. They obtained a high FOXP3⁄CD4 ratio in the symptomless skin of psoriatic patients [58]. Yan et al. detected an increase fraction in the psoriatic lesions irrespective of the severity of disease as compared to normal skin, but a decrease in FOXP3+cells from acute psoriatic biopsies. They suggested that FOXP3+Treg plays a crucial role in exacerbation of psoriasis not in a stable phase [56].
It is difficult to explain why we obtained a higher expression of CD4+ in perilesional skin than in psoriatic lesions. Moreover, CD4 is present not only on Tregs but is characteristic of Th1 and Th17. We expected to observe increasing CD4 in the lesional skin.
CD25(IL-2R) is present on activated T and B cells and used as a marker to identify CD4+FOXP3+Treg [59]. Most psoriasis-associated T cells are CD3+CD2+CD45RO+CLA+ with a subset having activation markers CD25, HLA-DR, and CD27 [60]. Therefore we reported a higher (p < 0.05) concentration of CD25/IL2R in control skin compared to psoriatic skin without any differences between the perilesional and lesional skin.
Duncan et al. demonstrated a significant correlation between the number of IL-2R+(CD25+) cells in lesional skin and PASI [61]. Similarly, Ferenczi et al. detected high numbers of IL-2R+T cells in lesional skin (epidermal and dermal layer) with an early activation of associated CD25 (α chain) molecule and with CD122 (β chain) [62].
CD39 expression has been described on FOXP3+Treg. It is the rate-limiting enzyme in the hydrolysis of extracellular ATP and ADP into AMP. Binding of adenosine leads to a rise of intracellular cAMP levels, which subsequently suppresses T effectors function such as proliferation and CD25 up-regulation [63]. The role of CD39+FOXP3+Treg has been investigated in multiple sclerosis: circulating CD39+FOXP3+Treg were diminished and impaired while CD39+FOXP3+Treg healthy individuals were suppressed by Th17 [64].
We did not find any differences in CD39 expression, contrary to others. They were scattered throughout the epidermis and dermis [19, 31, 56]. Zhang et al. assessed the proportions of CD39/CD73 expressing FOXP3+Treg in different types of psoriatic lesions and controls. In normal skin, CD39+cells were present and localized predominantly in the dermis, especially in the papillary and upper reticular layers of the dermis, and minimally in the epidermis. The proportion of cells in both CD39+ and FOXP3+ were significantly lower in pustular and erythrodermic psoriasis than in plaque psoriasis [30]. Similar results were reported by Bovenschen et al. and Yan et al. [19, 56]. However, they did not achieve a significant reduction in plaque psoriasis expression compared to pustular and erythrodermic psoriasis.
Tregs are characterized by low CD127(IL-7Rα) expression compared to conventional T cells. In our study, the expression of IL-7R/CD127 did not differ in analysed groups. Simonetta et al. investigated high CD127 expression on Treg in vitro and in vivo in contact dermatitis models. The opposite regulation of CD127 on Tregs and T effectors during activation led to equivalent CD127 expression on these two subsets. It is probably an additional mechanism for the functional impact of IL-17 on Treg suppression, which has been shown to rescue effector cells from Treg-mediated apoptosis [65].
CTLA4 is a critical negative regulator of T-cell activation and induces an inhibitory effect on B-cell inactivity, with a consequent decrease in autoantibody formation, decrease in macrophage activation, and reduction in pro-inflammatory cytokines [66, 67]. We observed higher (p < 0.05) CTLA-4 expression in psoriatic lesions, especially in the papillary layer.
The problem of our study is that the T effectors may exhibit transiently expressing CD25 and/or CTLA-4 in epidermis and dermis [18, 68]. Therefore Sugiyama et al. showed that epidermal and dermal cell suspensions rested for 48 h before flow cytometric analysis, because in CD4+CD25+Treg CTLA-4 is expressed up to 7 days after activation. They calculated the ratio of CD4+CD25+Treg vs. CD4+CD25− T effectors in psoriatic lesions to be 1 : 6 in dermis and 1 : 2 in epidermis [18].
Limitations of our research are a low number of patients and ambiguous identification of Tregs. Immunohistochemistry cannot provide convincing evidence for Treg identification.
Conclusions
The differences between the levels of protective cytokines and expression of Treg markers in perilesional skin and psoriatic lesions can be a proof of major disorders regarding the mechanisms of immune tolerance and might explain the inflammation development in psoriasis. Heterogeneous results of researches still suggest the need for further research on this topic.







