
Introduction
The latest breakthroughs in DNA-based technologies have contributed to the growth in exploration of bacterial gene functions and their role in human health. The objective of the Human Microbiome Project [1] is to study the diversity and function of the genes that reside in the human gut and other parts of the body. Scientists are particularly interested in how alteration in the intestinal microflora composition can affect the host’s health or development of diseases, particularly autoimmune diseases. The gut microbiome seems to have more than 3 million genes that produce thousands of metabolites, whereas the human genome has around 23,000 genes [2, 3]. The human gut microbiota is composed of four major phyla: Bacteroidetes (16%), Firmicutes (65%) Actinobacteria (9%), and Proteobacteria (5%) [4].
The term “dysbiosis” describes reduction in the microbial diversity caused by the loss of beneficial bacteria. It can also be triggered by the increase in the number of patients, which can become pathogenic. Imbalance in the microbial composition can be caused by three aspects: 1) loss of beneficial organisms, 2) excessive growth of potentially harmful organisms, and 3) loss of the overall microbial diversity [5].
The loss of diversity in the human gut is associated with various diseases, such as IBD and acute diarrhoea [6, 7]. Moreover, the gut microbiota’s immunomodulatory abilities are related to some dermatological diseases, such as psoriasis [8, 9] and atopic dermatitis [10, 11]. Not only can diseases affect changes in the diversity of the intestinal microbiota, but also some external factors, such as antibiotics [12, 13], heavy metals [14], or even artificial sweeteners [15, 16] can. The diversity of the gut microbiome is an ecological biomarker that can be used to assess ecosystem resilience [17].
Alopecia areata (AA) is a nonscarring hair loss disorder that affects nearly 2% of the general population with a wide spectrum of manifestations and an unpredictable course. It seems to be a T-cell-mediated autoimmune disease with an unknown etiopathogenesis. Skin biopsies of the AA -affected skin show a lymphocytic infiltrate in and around the bulb or lower part of the hair follicle in the anagen phase [18]. There is an intriguing link between AA and gut dysbiosis. However, although several studies demonstrated that the microbiome plays a critical role in the onset of skin diseases (including atopic dermatitis and psoriasis [12, 19–21]), the association between AA and gut dysbiosis awaits to be elucidated.
A recent study by Maslowski and Mackay [22] looked at microbiota as the basis for increased incidence of autoimmune conditions in developed countries. Short-chain fatty acids (SCFAs) produced by the gut microbiota contribute to maintaining immunological homeostasis via modulation of the regulatory T cells (Tregs) function and IL-10 release, which also play a critical role in the induction of AA [23].
To the best of our knowledge, only one study analyses the implication of the gut microbiome on the AA pathogenesis. This cross-sectional study involved 15 patients affected by alopecia universalis and 15 controls [24]. The gut microbiome of the study subjects was analysed by sequencing the 16SrRNA of the stool sample. In patients with alopecia, they found an enriched presence of Holdemania filiformis, Lachnospiraceae, Erysipelotrichacea, Clostridiales vadin BB60 group, and Parabacteroides distasonis. A predictive model based on the number of bacterial counts of Parabacteroides distasonis and Clostridiales vadin BB60 group correctly predicted a disease status in 80% of patients.
There is also 1 case report describing 2 patients with alopecia universalis who developed subsequent hair regrowth after faecal microbiota transplant (FMT) for treatment of recurrent Clostridium difficile infections, which may suggest the potential role of the gut microbiota in the mechanism of the disease [25, 26].
Aim
The aim of the study is to analyse the intestinal microbiome in patients suffering from alopecia areata.
Material and methods
This study was conducted at the Medical University of Lodz; Department of Dermatology, Paediatric, and Oncology and was supported by “Operation Integration!” Integrated Program of the Medical University of Lodz (Grant No. POWR.03.05.00-00-z065/17). Based on the methodology described below, this study should be considered as a pilot study that requires further evaluation. It included 25 patients above 18 years of age with AA who were hospitalized at the Department of Dermatology, Paediatric, and Oncology. Age, sex, duration of the disease, and any systemic or autoimmune diseases were being recorded following the special “Investigative guidelines for AA “, while the scalp involvement was assessed using the Severity of Alopecia Tool (SALT) [27]. The inclusion criteria included patients diagnosed with AA and an active disease state when the stool sample was collected within a month. The exclusion criteria were as follows: 1) the patients who underwent either a systemic immunosuppressant therapy or treatment with glucocorticoids or dithranol before hospitalization; 2) patients with signs of infection like fever, sore throat, cough, or dysuria; 3) patients who took antibiotics within 30 days or probiotics within 15 days before the sampling. The participants were classified into the following groups: 1) AA patients with < 25% scalp involvement; 2) AA patients with 25–99% scalp involvement; 3) patients with alopecia totalis (AT) or alopecia universalis (AU). Written consent was obtained from all the subjects prior to their participation in the study. The participants in the study were instructed to take a sample of their first faeces in the morning and place it in sterile plastic containers. The samples were stored in the Isohelix StoolFix faecal microbiome DNA stabilization kit at room temperature according to the manufacturer’s instructions. Negative controls involved performing the extraction without any clinical sample. The study was carried out in compliance with the Declaration of Helsinki principles and was reviewed and approved by the Bioethics Committee of the Medical University of Lodz, Poland.
Our study involved 25 people previously diagnosed with patch alopecia, of which 19 were women and 6 were men. Their average age was 36.8 years (95% CI: 29.4–47.8) and co-morbidities included atopic dermatitis, psoriasis, autoimmune thyroiditis, vitiligo, rheumatologic arthritis, and type 1 diabetes. The demographic details are presented in Table 1. Here, we examined the human gut microbial community at the species level by metataxonomics. To achieve this purpose, a high-throughput approach involving operational phylogenetic unit analysis of the nearly full-length 16S ribosomal RNA (rRNA) gene sequence was used. The DNA from stool samples was isolated using a QIAamp DNA Microbiome Kit with modifications to increase DNA concentration. The isolated DNA from each faecal content sample was used to construct a sequencing library targeting the V3–V4 region of the bacterial 16S rRNA gene. Overhang adapter sequences were appended to the primer pair for compatibility with Illumina index and sequencing adapters. Negative controls were amplified for all barcode-primer sets. Then, each sample was amplified using a limited cycle PCR program, adding Illumina sequencing adapters and dual-index barcodes to the amplicon target. The final libraries were again purified, quantified, and normalized prior to pooling. Automated cluster generation and paired-end sequencing with dual reads were performed according to the manufacturer’s instructions. The clustered sequences were utilized to construct Operational Taxonomic Units (OTUs) tables with 97% identity and representative sequences were classified into the respective taxonomical level from phylum to genus based on the Greengenes 16S rRNA gene database.
Table 1
Demographic details of the AA donors – sex, age, comorbidities, SALT index, assessed group based on SALT index and most abundant family
Statistical analysis
All statistical analyses were performed with MicrobiomeAnalyst [28]. The overrepresentation of bacterial taxa was tested statistically at the family and genus level. P-values were converted to false discovery rate values (FDR) to correct for multiple testing, p ≤ 0.05 was considered statistically significant. A false discovery rate (FDR) of less than 0.1 was considered significant for multiple comparisons. Multiple longitudinal assessments of a given Genera or Family among participants from two different groups were analysed using ANOVA as we were able to perform a univariate analysis. The Shannon diversity index was used to assess biodiversity of a sample and abundance of organisms. In essence, this test considers the number of species and their evenness. Statistical differences in this index were determined using the Mann-Whitney U test. Alfa and beta diversity was used to measure and compare the diversity or composition of microbial communities within samples. The observed indexes calculate the actual number of unique taxa observed in each sample, estimates the changes in the presence/absence of thousands of taxa in a dataset and summarises them by stating how similar or dissimilar two samples are [29, 30]. By exploiting a Principal Coordinate Analysis (PcoA), we visualise a matrix in the 2D plot where each plot represents the entire microbiome of a single sample. The hierarchical clustering result is shown as a heatmap to identify relationships among various data sets where data values are transformed to the colour scale [31]. The microbial composition at the family level was determined by performing a univariate analysis. The features are significant based on their adjusted p-value, the default is p-value cutoff = 0.05.
Results
Before data analysis, an integrity check was performed to ensure that all the necessary details were collected. Figure 1 shows the library size for the inspection of each sample. The assessment of the library range fluctuates from 975 to 64218 bp. It shows the taxonomic composition using a stacked bar/area plot. Histograms were based on the abundance on taxonomic levels like family and genus. The genetic material of bacteria having the lowest abundance in samples was unified to a non-assigned group. The bacterial families found in all 25 samples are Lachnospiraceae, Enterobacteriaceae, Bifidobacteriaceae, Streptococcaceae, Eubacteriaceae, Ruminococcaceae, Eggerthellaceae, Peptostreptococcaceae, and Clostridiaceae. This causes the domination of 3 bacteria phyla: Firmicutes, Proteobacteria, Actinobacteria, with a significant domination of Firmicutes phylum. Apart from the KA4 sample, the Lachnospiraceae family was the most abundant in the rest of the samples. Figure 1 shows the taxonomic composition of the community at the family level. Apart from the above-mentioned groups, a large part of the histograms is occupied by the genetic material of bacteria not assigned to the dominant groups - collectively they have been described as “not assigned”.
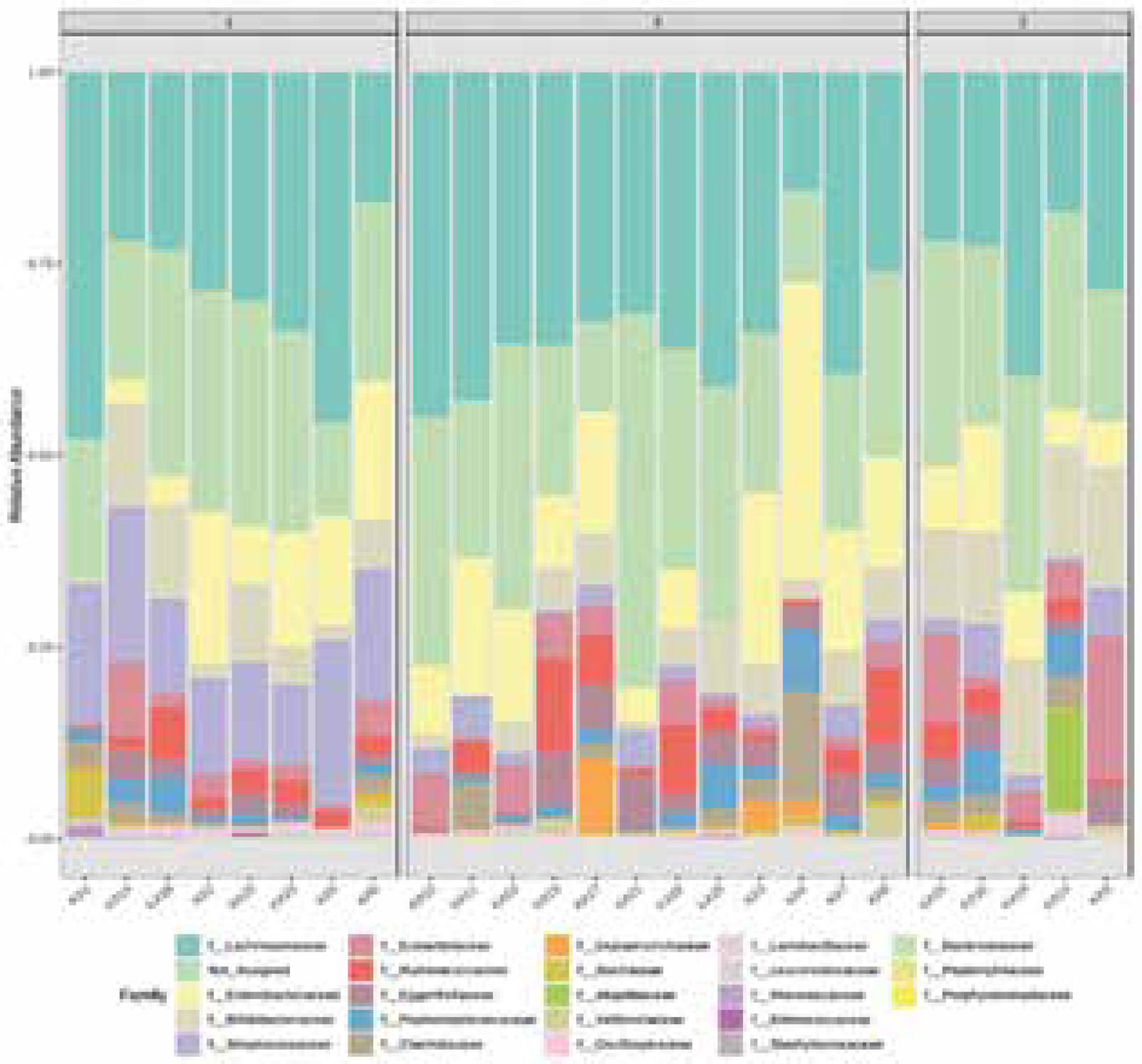
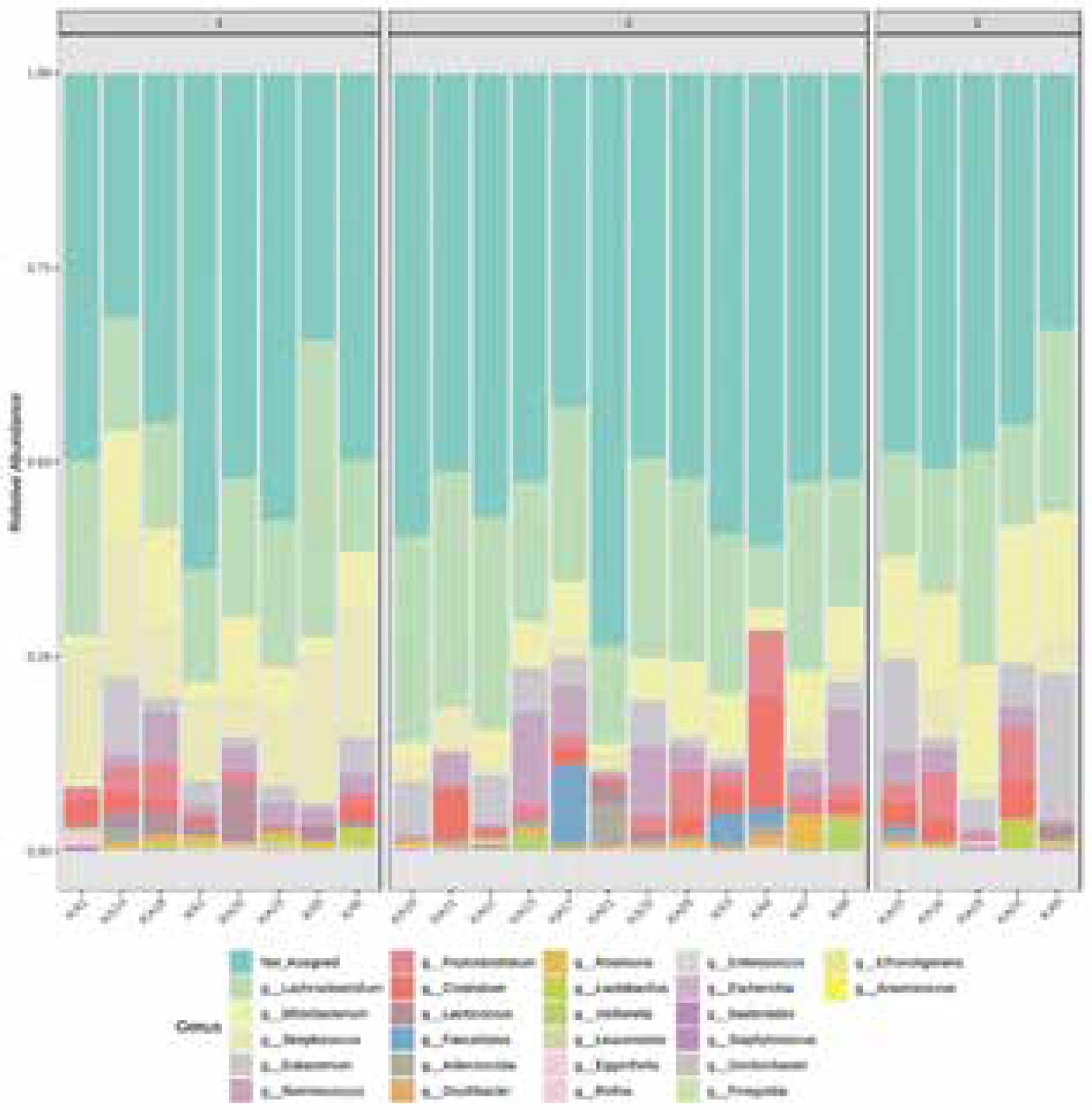
Figure 2 reveals the taxonomic composition of the community at the genus level. Lachnoclostridium, Bifidobacterium, Streptococcus, Eubacterium, Ruminococcus, Peptoclostridium were detected in all the samples in different proportions. Clostridium was found in eighteen samples and it was the dominant genus in sample KA4. Lactococcus was present in thirteen samples, with the highest abundance in KA20. Faecalitalea was conspicuous in seven samples. Adlercreutzia was observed in ten samples. Roseburia was found in six samples. Lactobacillus, in only a few percentages of microbiome composition, was present in fifteen samples. In the analysed samples other genera occurred at < 1% abundance Bacteroides.
Alpha diversity and B-diversity analysis
We used Alpha-diversity to calculate richness and evenness within each of the 25 samples. Figure 3 shows the alpha diversity measure across samples for a given diversity index. Ka14 had the highest alpha diversity while Ka16 had the lowest. Figure 4 shows the diversity distribution using a box plot of a group present within the Group taxonomy class, statistical significance p-value 0.0215, F value 4.59.
Figure 3
Alpha-diversity at the family level across all samples. The samples all represented on X-axis and their estimated diversity on Y-axis
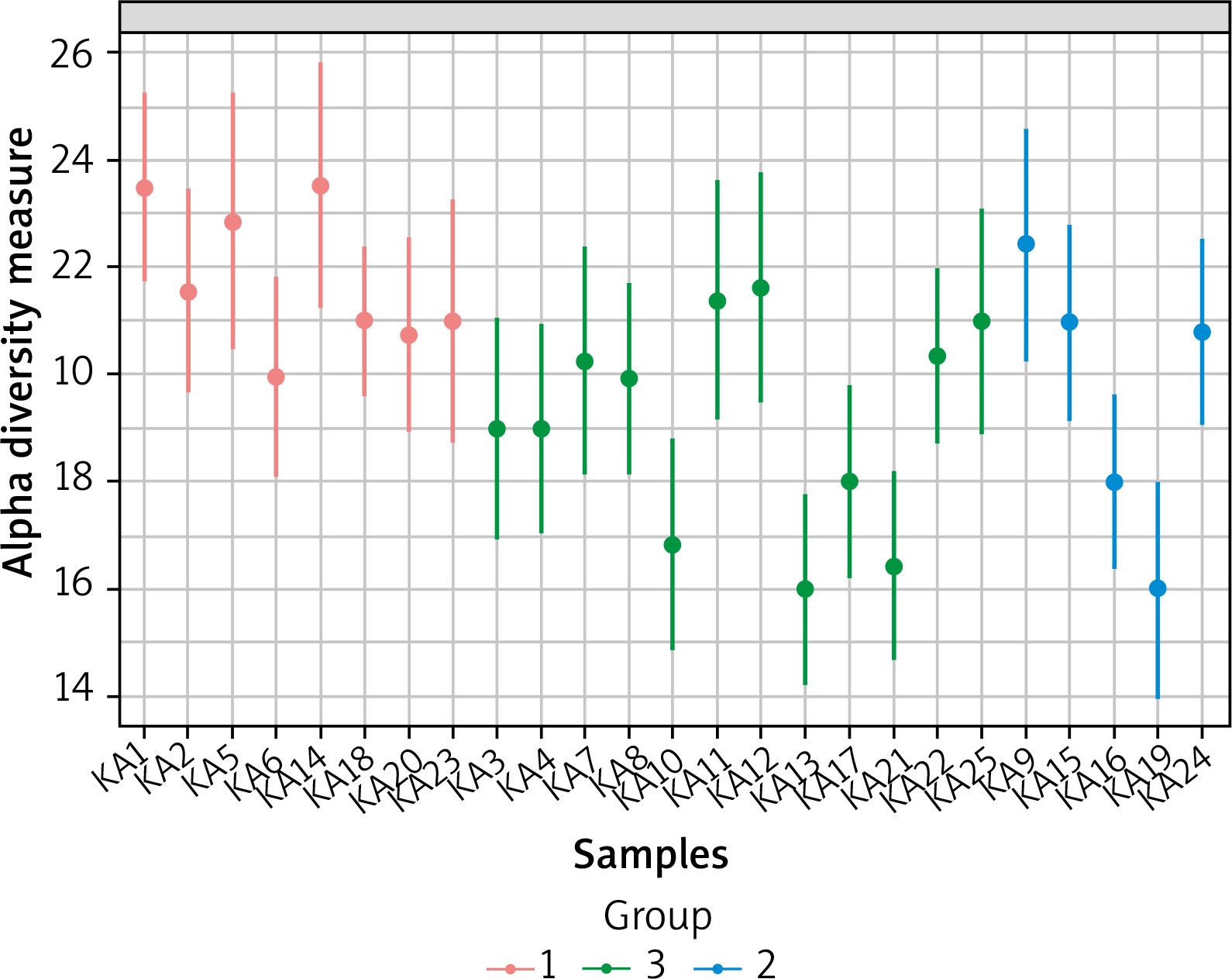
Figure 4
2-D Principle coordinate analysis (PcoA) plot using bray distance. Statistical significance (PERMANOVA) F-value: 3.6451; p-value < 0.001
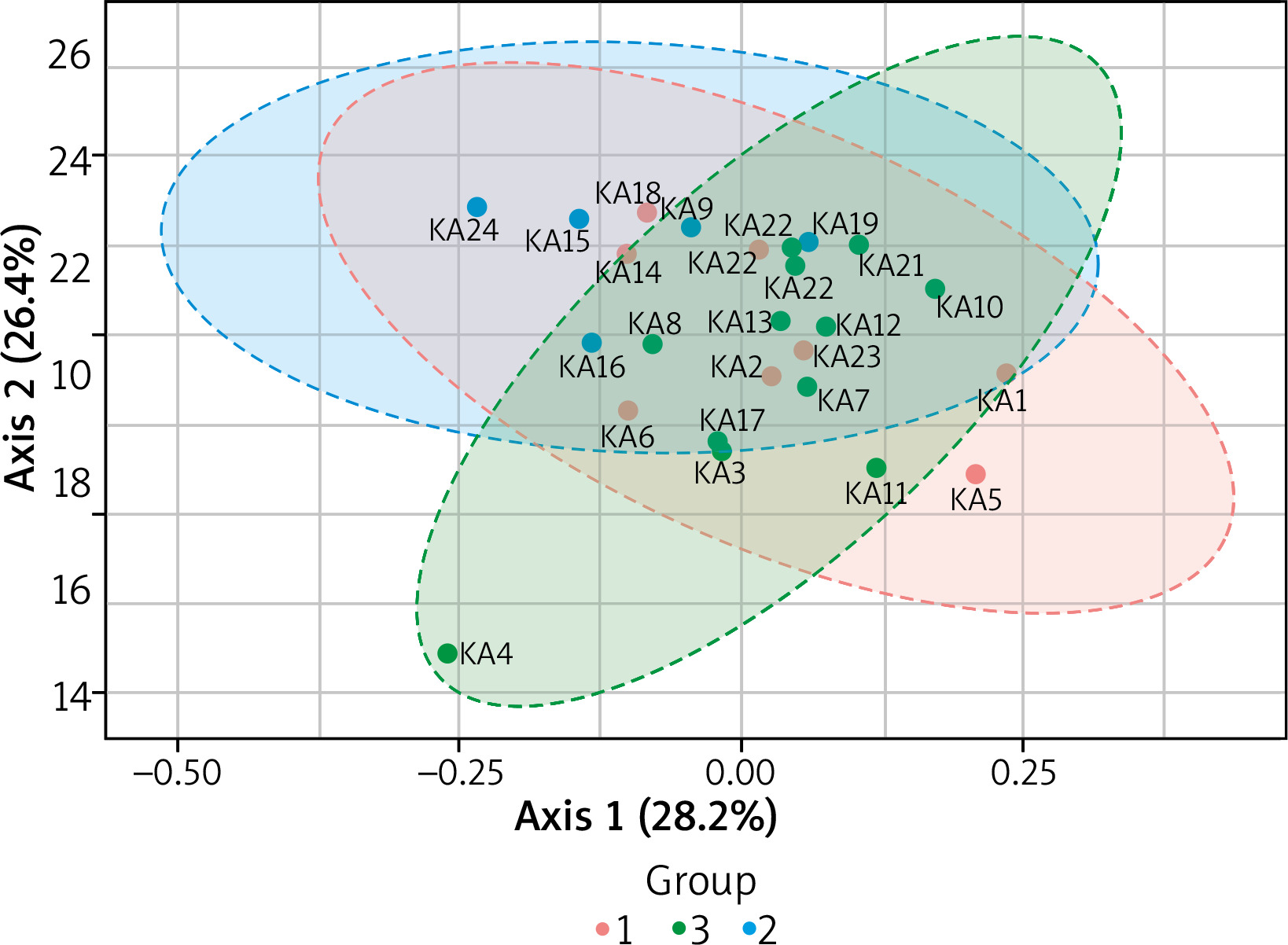
To compare composition of microbial communities we use a beta diversity analysis and Principal Coordinate Analysis (PcoA) to visualise the results. A statistical significance is evaluated using the ANOVA F-value: 3.645; R2 = 0.248; p-value < 0.001. Figure 4 shows the diversity analysis by a PcoA plot using Bray distance. KA4, KA24, KA18, KA5 samples did not cluster together indicating their different beta diversity.
The clustering result is shown as a heatmap
Our hierarchical visualization shows that the majority of families occur in low abundance. Only a few samples contained a larger amount of one family of bacteria. Group 1 is characterized by a higher presence of the Streptococcaceae family, and group 3, compared to the rest of the samples, has a higher abundance of Bifidobacteriaceae. Except for the isolated family bacteria in the samples described, the core of the analysed microbiome is low diverse.
To describe the intestinal bacterial composition in the AA population, we use a heatmap representing the core microbiome at the genus (Figure 5) and family level. Microbiome core analysis constitutes Lachnoclostridium, Bifidobacterium, Streptococcus, Eubacterium, Ruminococcus, Peptoclostridium, and Clostridium, and, to conclude, almost all of them (aside from Bifidobacterium) belong to the Firmicutes phylum. Besides the dominance of the Firmicutes, the other phyla found in higher proportions in our samples are Actinobacteria and Proteobacteria.
Univariate analysis
Out of all the microbiome data identified in the analysis, only four recognized bacterial families meet the statistical criteria described above. Streptococcaceae, Bidobacteriaceae, Leuconostocaceae, and Eubacteriaceae achieve p-value below 5%. That gives us a small number of properly identified microbiome, as compared to the high number of features surviving statistical criteria. Retaining those significant features with higher FC values for the AA intestinal microbiome analysis requires follow-up validation studies.
Discussion
In our study, we wanted to analyse the composition of the intestinal microbiome of the patients suffering from AA and confront the obtained results with the emerging speculations about the influence of the intestinal dysbiosis on the pathogenesis of AA. This is one of the first analyses of the microbiome core in AA patients and we would like to emphasize that it should only be an introduction to the search for correlation between the intestinal dysbiosis and the etiopathogenesis of AA.
Bacteroides is a Gram-negative microflora in the human intestinal tract, which together with the Firmicutes phyla accounts for 90% of the intestinal bacterial composition [32]. Bacteroides have a symbiotic host-bacterial relationship with humans. They assist in breaking down food and producing valuable nutrients and energy like polysaccharide A [33]. The mechanisms of immunological regulation mediated by Bacteroides fragilis have been extensively studied. B. fragilis capsular polysaccharide A (PSA) activates Treg cells via TLR2 produced by Foxp3+Treg, which emit the anti-inflammatory cytokine-10, preventing inflammation in the gut [34, 35].
Nowadays some interesting studies link lower abundance of Bacteroides in the intestinal composition and disruptions in the gut symbiosis with the pathogenesis of many diseases, for example, inflammatory bowel disease. The findings of low Bacteroides levels in patients with IBD are supported by the meta-analysis conducted by Zhou and Zhi [36]. Despite the heterogeneity in methodologies, the meta-analysis found that IBD patients with active illness had low levels of Bacteroides [36]. In the literature, there has been a greater focus on the co-morbidity of IBD and AA. The absence of hair in the body areas where it normally grows, which is indicative of AA, is a quite common symptom seen in IBD patients in clinical practice. The prevalence of AA in Colitis Ulcerosa (UC) patients is much higher than in the general population (1 : 47 vs. 1 : 125–1 : 2439) [37]. Several studies have found a relationship between AA and UC, and a link between genetic susceptibility, family aggregation, and HLA has been suggested [37].
Sobolewska-Wlodarczyk et al. [38] recently published an analysis on co-morbidity between AA and IBD (inflammatory bowel disease) concluding that the pathophysiology of AA occurring in the IBD patients is unknown. Hair loss, which is typical of AA, is seen in the IBD patients in the clinical practice, although it often goes unreported. As a result, the IBD patients need to keep a close eye on their skin and work with a dermatologist. Based on the conclusions, the low abundance of Bacteroides in the intestinal microbiome of the AA patients needs to be evaluated based on a higher number of clinical trials.
The Firmicutes phylum is observed to be enriched in our samples primarily due to the enrichment of the Lachnospiraceae family. Out of 25 samples, 23 included the Lachnospiraceae family, the highest percentage of the identified abundance in the samples. The Lachnospiraceae family is obligatory anaerobic constituents of the microbiota [39, 40]. They influence their hosts by creating short-chain fatty acids, small molecules that accumulate in the colon and circulate systemically, particularly butyrate, acetate, and propionate [41–43]. SCFAs influence the microbial environment and interact directly with the immune system of the host. Moreover, the existence of SCFAs also results in enhanced histone epigenetic states in the host and lower levels of inflammatory markers. Furthermore, the number of colonic regulatory T cells (Tregs), which are necessary for self-antigen tolerance and the prevention of autoimmune illness, has decreased [44].
The Lachnospiraceae family has a profoundly positive impact on the host’s health, maintaining the gut microbiome balance and improving intestinal epithelial integrity [40]. Additionally, in some recent studies, a pathologic state, like inflammatory bowel disease, was correlated with lower amounts of Lachnospiraceae in UC faecal samples [45]. On the other hand, Ley et al. [46] described a statistically significant dependence between the increased abundance of Firmicutes and decrease of Bacteroidetes associated with a high body mass index (BMI) [47, 48]. The effect of the increased abundance of Firmicutes in comparison to the Bacteroides level has been described as the Firmicutes to Bacteroidetes ratio (F/B ratio), which has been extensively examined for human and mouse gut microbiota. Stojanov et al. wrote “Dysbiosis is defined as an increase or reduction in the F/B ratio, with the former usually observed with obesity, and the latter with inflammatory bowel disease (IBD)” [49]. Recently, the B/F ratio was also used in some dermatological diseases.
This indicator has been lately described within the context of the intestinal microbiome in dermatological diseases such as psoriasis. Shapiro et al. [50] described a dominance of the Firmicutes phylum and a marked increase in Actinobacteria prevalence in psoriasis patients. Dei-Cas et al. [51] and Hidalgo-Cantabrana et al. [52] drew similar conclusions from their metagenomic meta-analysis of the gut microbiota. According to these studies, the psoriasis microbiome was found to have a high Firmicutes/Bacteroidetes ratio.
The only research that investigates the proportions of the gut macrobiotic composition in patients with AA that we have found is the study by Margit [53], who reported a statistically significant decrease in Bacteroides and increase in the Firmicutes quantity. However, we must admit that determination of the intestinal dysbiosis, based on a change in the proportion of the Bacteroides to Firmicutes, and therefore its impact on dermatological diseases, requires extensive research. This is due to numerous factors influencing the proportions of the intestinal microflora like diet, age, ethnicity, physical activity, food additives and contaminants, antibiotic consumption [54, 55].
The dysbiotic gut microbiome is also featured as a lower proportion of anaerobes that normally predominate in healthy subjects to a higher proportion of facultative anaerobes, such as Proteobacteria and Bacilli [56]. Loss of health-associated strict anaerobes can be caused by a variety of circumstances in various disorders, including the use of broad-spectrum antibiotics, a breakdown in host-microbe interactions promoted by host genetics, or other factors [57, 58]. Therefore, we took into consideration the proportions between anaerobic bacteria and facultative anaerobes in our samples. The microbiome analysed by us is in majority composed of an anaerobic genus. Thus, we cannot describe bacterial dysbiosis understood in the context of an increased proportion of facultative anaerobes.
In the present study, the four main genera that constitute the core of the microbiome are Lachnoclostridium, Bifidobacterium, Streptococcus, Eubacterium, and 3 main phyla, namely Firmicutes, Proteobacteria and Actinobacteria. The other genera and phyla were found in a negligible proportion. In samples, the representation of bacteria of the phylum Bacteroidetes was reduced. The overrepresentation of Firmicutes and Proteobacteria may indicate a disturbed microflora [59]. Additionally, the composition of bacterial communities reveals an overall loss of richness in all samples and a decrease of taxonomic diversity.
The main limitation of this study is a multitude of factors that may affect changes in the composition of the gut microbiota, ranging from the participants’ age, to their ethnicity, lifestyle, eating habits, or coexisting diseases. Moreover, due to the influence of ethnicity and geographical origin on the gut microbiome composition, it also proves challenging to find the right formula to reliably compare data from available publications. We classify this research as a pilot study because our analysis requires confirmation of the findings of healthy individuals from the same ethnic group, preferably family members of AA patients.
Conclusions
Overall, the information presented in our study helps us to understand the importance of further work on the analysis of the relationship between dermatological diseases, especially AA, and the disturbance of the intestinal microbiome diversity. Despite the disturbances in the analysed intestinal microbiome of the AA patients, to confirm their relationship with the AA etiopathogenesis more investigation needs to be performed.







