
Introduction
Melanoma derives from melanocytes localised in the skin (approximately 90%) or mucosa. Approximately 40% of skin melanomas come from pre-existing nevi. Development of this cancer is determined by genetic factors such as presence of numerous and/or dysplastic nevi, familial history of melanoma, skin phenotype, and environmental such as UV exposure [1].
Melanoma is one of the most aggressive cancers with an unpredictable course of disease. It accounts for 4% of all skin cancers, but is responsible for 75% of deaths caused by these malignancies [2]. Its aggressiveness is manifested by potent invasiveness and high metastatic potential. Even a tumour of < 2 mm thickness can lead to metastasis. Due to early detection the mortality rate in Australia and the USA is relatively low (10%) [3]. However, approximately 40% of melanoma patients in Poland still die from this disease (Polish National Registry of Cancers, 2015). Since advanced melanoma is radio- and chemoresistant at this stage the disease is still related to poor prognosis with reported 5-year survival rates at a level of 10% [4]. Until 2016 there was no efficient adjuvant therapy for patients at high risk of metastatic disease and until 2011 there were no drugs prolonging survival of advanced patients. A significant improvement in melanoma treatment has been made due to substantial progress in the understanding of melanoma biology. Since 2011 eight new drugs have been registered for advanced melanoma treatment. They constitute both molecular targeted therapies and immunotherapy. The first category consists of small molecules targeting the most important signalling pathway in melanoma, mitogen-activated protein kinases (MAPK) pathway. The second is antibodies which restore immune response or genetically modified viruses which stimulate the immune system against melanoma [4].
From melanoma initiation to radial growth phase
The prerequisite for melanoma development is the uncontrolled proliferation of melanocytes and melanoma cells. Most cancers acquire this ability by constant activation of signalling pathways regulating this process. In melanoma the proliferation signal goes mainly through MAPK signalling (Ras/Raf/MEK/ERK), which is activated in approximately 80–90% of melanoma cases. This pathway also regulates such tumour-promoting processes as growth, invasion, angiogenesis or resistance to therapy [5, 6]. In melanoma, the MAPK pathway is activated mainly by mutation in B-Raf proto-oncogene (BRAF) and to a lesser extent by mutations in N-Ras proto-oncogene (NRAS) (15–30%) and KIT proto-oncogene receptor tyrosine kinase (c-KIT) (2–5%) [5, 7]. The predominant mutation in BRAF is the substitution of valine with glutamic acid at codon 600 (BRAFV600E) which leads to a huge increase in BRAF activation, 700 times exceeding that of the wild form [8]. BRAF mutation is detected in approximately 50% of cases and is the most common genetic change in melanoma. The high frequency of this mutation and “addiction” of melanoma cells to activated BRAF makes it an excellent therapeutic target. So far two drugs targeting BRAF (vemurafenib and dabrafenib) were accepted by the Food and Drug Administration (FDA) and are standard therapies for advanced melanoma patients with BRAF mutation [9]. More than 50% of patients respond to vemurafenib and some of them experience a complete response. Similar results were obtained for dabrafenib with a 60% response. The response to BRAF inhibitors is very fast and spectacular. However, it does not last long and the patients’ survival is not prolonged significantly. The median overall survival (OS) of patients treated with targeted therapy is 14 months while for dacarbazine it is 9 months. Fast relapse after therapy completion stems from regrowth of melanoma due to intrinsic or acquired resistance to BRAF inhibitors [10]. To overcome the resistance, another MAPK pathway inhibitor, trametinib (MEK inhibitor) was approved in 2013. The new drug shows a worse response rate than the previous drugs, but in contrast to BRAF inhibitors, no cutaneous squamous cell carcinomas or hyperproliferative skin lesions appear during treatment. Additionally, combination of trametinib and dabrafenib showed a promise in delaying the development of resistance to MAPK inhibitors [11]. The combination of BRAF and MEK inhibitors (dabrafenib and trametinib or vemurafenib and cobimetinib) shows significantly longer progression-free survival (PFS) and OS as compared to BRAF inhibitors monotherapy (with median OS exceeding 2 years; for more clinical data, Table 1 A) [12, 13]. Also, a combination of encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) prolongs PFS of advanced melanoma patients as compared to monotherapy [14]. This combination has already been approved for melanoma treatment. Although the list of anti-melanoma targeted drugs is getting longer, the intrinsic or acquired resistance to BRAF and MEK inhibitors is still a challenge in melanoma treatment. A substantial amount of data was gathered on this subject, which is beyond the scope of this article, however.
Table 1
Approved or recommended therapies for adjuvant or advanced melanoma treatment – selected clinical results and future perspectives. For more detailed data on clinical trials see review papers [4, 11, 59, 93]
| A. Drugs and therapies | |||||
|---|---|---|---|---|---|
| Drug(s) | Target | Objective response rate (ORR) | Median OS | Most common adverse effects (AE) | References |
| Chemotherapy: | |||||
| Hydroxyurea | RNR (ribonucleotide reductase) | 20%, (+ radio-therapy) | – | Bone marrow depression, gastrointestinal symptoms, dermatological reactions | [4] |
| Dacarbazine (DTIC) | DNA alkylation | 5–20% | No benefit | Anorexia, nausea, and vomiting | [4] |
| Fotemustine | DNA alkylation | 15, 2% | 7.3 months vs. 5.6 months (DTIC) | Neutropenia, thrombocytopenia | [79] |
| Targeted therapy: | |||||
| Vemurafenib (BRIM-3 study) | BRAF | 57% | 13.7 months vs. 9.7 months (DTIC) | Cutaneous squamous cell carcinomas/keratoacanthomas, rash, abnormal liver function | [80] |
| Dabrafenib (BREAK-2 study) | BRAF | 59% | 13.2 months | Hyperkeratosis, papillomas, hand-foot syndrome, squamous cell carcinomas/keratoacanthomas | [81] |
| Trametinib | MEK | 25% | 14.2 months | Rash, diarrhoea, peripheral oedema, pruritus, fatigue | [82] |
| Dabrafenib + Trametinib | BRAF, MEK | 69% | 25.1 vs. 18.7 months (dabrafenib) | Pyrexia, chills, diarrhoea, hyperkeratosis, vomiting, peripheral oedema | [13, 83] |
| Vemurafenib + Cobimetinib (coBRIM study) | BRAF, MEK | 70% | 22.3 vs. 17.4 months (vemurafenib) | Nausea, vomiting, and diarrhoea, creatine kinase level, retinopathy | [12] |
| Immunotherapy: | |||||
| Interleukin-2 (IL-2) | Stimulation of immune system | 16% | 11.4 months | Hypotension, nausea, vomiting, diarrhoea, confusion, oliguria, thrombocytopenia, fever | [84] |
| Ipilimumab | CTLA-4 | 23–32.6% | 8.5––14.3 months | Enterocolitis, hepatitis, dermatitis, diarrhoea, fatigue | [85] |
| Nivolumab | PD-1 | 40% | Not reached (41% > 3 years) | Pneumonitis, fatigue, rash, pruritus, diarrhoea, constipation, asthenia, vitiligo | [76] |
| Pembrolizumab | PD-1 | 33% | Not reached (74% – 1 year survival) | Fatigue, rash, pruritus, diarrhoea, asthenia, vitiligo, nausea, arthralgia | [86] |
| Ipilimumab + Nivolumab | CTLA-4, PD-1 | 58% | Not reached 3 years survival: 58% (nivolumab-plus-ipilimumab), 52% (nivolumab), 34% (ipilimumab) | Rash, pruritus, fatigue, diarrhoea, nausea, vitiligo, hypothyroidism and many others | [87] |
| Talimogene laherparepvec (T-Vec) | Local oncolysis, stimulation of immune system | 26.4% | 23.3 vs. 19 months (GM-CSF) | Flu-like syndrome, pyrexia, chills, fatigue, malaise, nausea, localized pain, headache, vitiligo | [80] |
| Adjuvant therapies: | |||||
| Interferon α2b | Stimulation of immune system | n.a. | 3.8 vs. 2.8 years | Fever, chill, flew-like symptoms, myelosuppression, hepatotoxicity, neurologic AE | [88] |
| Peginterferon α2b | n.a. | RFS: 34.8 vs. 25.5 months | Fatigue, increased alanine aminotransferase (ALT) and aspartate aminotransferase (AST), pyrexia, headache, anorexia, myalgia, nausea, chills, and injection site reactions | [89] | |
| Ipilimumab | RFS: 26.1 vs. 17.1 months | Rash, pruritus, fatigue, diarrhoea, nausea, abdominal pain, vomiting, headache, weight loss | [63] | ||
| Other therapies not approved by the FDA: | |||||
| Imiquimod | Stimulation of immune system | Recommended as treatment options for patients with in-transit melanoma metastases | [90] | ||
| B. Emerging targets and drugs | |||||
| Encorafenib, binimetinib, masitinib | BRAF, MEK, C-KIT, respectively | [14] | |||
| Epacadostat, Indoximod | IDO1 (indoleamine 2,3-dioxygenase 1) | [91] | |||
| Antagonists of other inhibitory receptors on T cells | TIM-3, LAG-3 TIGIT, BTLA | ||||
| Agonists of costimulatory receptors on T cells | CD27 (Varlilumab), OX40, GITR | ||||
| Entinostat | HDAC | ||||
| LEE011 | CDK4/CDK6 | [45] | |||
| SAR245409 | PI3K/mTOR | ||||
| BYL719 | PI3K | ||||
| Oncolytic therapies | PV10 (10% Rose Bengal disodium) T-pIL12-EP (DNA plasmid encoding interleukin-12 CAVATAK (oncolytic strain of Coxsackie virus A21) HF10 (nonselective clone from the non-neuroinvasive HSV-1 strain HF) | [92] | |||
| For various combination therapies see [45] | |||||
Mutated BRAF highly stimulates melanoma growth but is not sufficient to promote melanoma development on its own and the vast majority (80%) of benign nevi also harbour this genetic alteration [6].
BRAF and NRAS mutations exclude each other, but are responsible for MAPK pathway activation in almost 90% of cases [5,6]. In rare types of melanoma the most common oncogene which sustains melanoma proliferation is c-KIT which is mutated in 39% of mucosal melanoma, 36% cases of acral melanoma and 28% of the lentigo type [15, 16]. In these cases a response to imatinib [17], and nilotinib [18], selective inhibitors of tyrosine kinases including c-KIT, is observed.
Activation of MAPK pathway due to mutations either in BRAF, NRAS or c-KIT initiates the neoplastic transformation by sustaining melanocytes/melanoma proliferation. However, activation of oncogene usually leads to oncogene-induced senescence [19]. That is why additional genetic changes have to occur to prevent senescence and complete the transformation process. The most common is mutation in the promotor of TERT gene (telomerase reverse transcriptase). TERT maintains telomere ends and its activity in cancer cells [20] prevents their senescence. Approximately 70% of sporadic melanomas harbour mutation in the promotor of this gene leading to a few times higher expression of TERT. This mutation is induced by UV probably at the initial stage of melanoma development as it is present in most melanoma intermediate lesions and melanoma in situ [21, 22]. Also, heritable mutations in TERT and genes associated with telomerase activity (POT1, TERF2IP, ACD) were described. These genetic changes predispose to melanoma at the early age [23, 24] by preventing cellular senescence.
Melanomagenesis is also driven by activation of the NOTCH pathway. NOTCH-1- NOTCH-4 proteins are transmembrane receptors activated by transmembrane ligands present on neighbouring cells. They regulate differentiation and renewal of stem cells. Their altered expression is observed in various tumour types including melanomas [25]. Activated NOTCH is also present in dysplastic nevi where it facilitates the anchorage-independent growth of melanocytes. In melanoma NOTCH proteins promote invasiveness, vasculogenic mimicry [26] and angiogenesis [27]. Inhibition of this pathway leads to apoptosis specifically in melanoma cells but not melanocytes, which is very promising in terms of potential anti-NOTCH therapy. On the other hand, NOTCH proteins may also act as tumour suppressors, e.g. NOTCH-3 [28] and NOTCH-4 [29]. A complex role of NOTCH proteins makes this pathway a challenging target for therapy. So far inhibitors of NOTCH have shown minimal clinical activity as a single agent [30]. However, some studies suggest that targeting NOTCH may be clinically beneficial in combination with other targeted drugs such as ERK [31] or ERBB inhibitors [32].
Along with pathways regulating proliferation and senescence, those controlling differentiation of melanocytes are also involved in malignant transformation e.g. melanogenesis associated transcription factor (MITF) pathway. MITF is a transcription factor required for development of melanocyte lineage from the neural crest. Its shortest isoform codes MITF-M, specifically expressed in melanocytes. It controls melanin synthesis, melanosomes production, differentiation, growth and survival [33]. In 2005 MITF was identified as a melanoma oncogene amplified in 10–20% of melanoma metastasis [34]. Point mutations in MITF or SOX10 (transcription factor acting upstream of MITF signalling) are also present in primary melanomas [35]. MITF expression at an optimal level seems crucial for sustained proliferation and survival of melanoma cells. It was observed that a low level of MITF leads to growth suppression, while a too high amount of this protein induces differentiation [5, 7]. Interestingly, BRAF oncogene probably regulates the optimal expression of MITF by its stimulation on the one hand and degradation on the other [36]. Cooperation of those two oncogenes seems crucial for melanocyte transformation [5, 7]. In vitro results suggest that inhibiting MITF may suppress melanoma growth [37], but the nonlinear relationship of its activity and expression makes it a rather difficult therapeutic target.
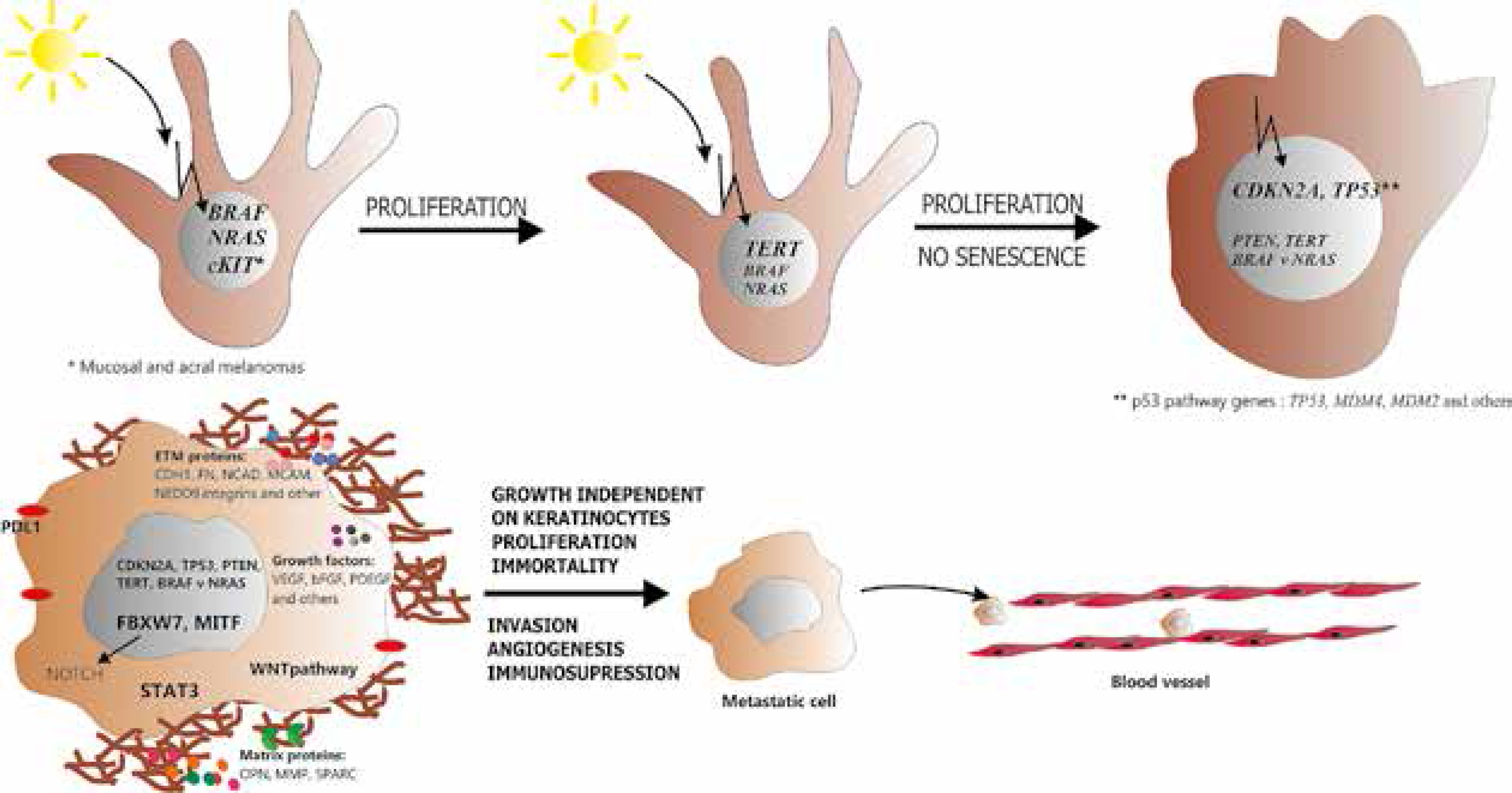
To sum up, activation of BRAF, NRAS and NOTCH leads to uncontrolled proliferation of melanocytes which may give rise to benign naevus. Upon TERT overexpression or suppression of RB and/or p53 pathways the senescence is blocked and the naevus turns into melanoma in the radial growth phase.
Progression – vertical growth phase
Vertical growth phase (VGP) melanoma is characterised by the presence of a dominant nest within the papillary dermis, cells with a distinct cytomorphology and mitosis [38]. The most significant and clinically relevant features of VGP melanoma are uncontrolled proliferation and capacity for invasion and metastasis.
The first one is acquired by mutations in genes regulating the cell cycle and cell death. There are two main tumour suppressors involved: RB and p53. RB is one of the most important negative regulators of the cell cycle. Its impaired activity leads to uncontrolled cell divisions. RB is mutated in 14% of melanomas [39], but its activity is suppressed in a vast majority of melanomas due to lack of expression of its activator protein p16. p16 is coded by CDKN2A (cyclin dependent kinase inhibitor 2A) gene mutated in 40–50% of melanoma cases [40, 41]. Inactivation or low expression of p16 may result from epigenetic modifications (mainly hypermethylation of its promoter) [42], expression of its inhibitor, ID1, and activation of WNT/β-catenin pathway [43]. p16 protein is an RB activator and protects the cell from malignant transformation initiated by BRAF or NRAS mutations. It activates RB protein by binding and inhibiting cyclin dependent kinase 4 (CDK4) [44] thus inhibition of CDK4 is being tested as a potential new targeted therapy for advanced melanoma [45].
CDKN2A is a very important tumour suppressor since along with p16 (P16INK4A) it also encodes p14 protein (P14ARF). While p16 regulates RB pathway, p14 regulates p53 pathway. Consequently, CDKN2A mutations may simultaneously impair two melanoma suppressors. That is why CDKN2A mutations are the most common heritable aberrations in familial melanoma [46].
Contrary to other neoplasms, mutations in tumour protein 53 (TP53) are relatively rare in melanoma with a frequency of 17% [41]. However, inhibition of p53 pathway is more common due to mutations in regulators of p53 such as MDM2, MDM4, CDKN2A(ARF) and others (ASPP, TP73, TP63) [41]. An increased amount of MDM4 for example is observed in almost 60% of melanoma cases [47]. Inactivation of RB and p53 pathways is present in 90% of melanoma cell lines and together with BRAF [48] or NRAS mutations contributes to melanoma development [49].
Some melanoma researchers suggest that activation of MAPK pathway due to BRAF or NRAS mutations is responsible for initiation of melanocytes transformation while further development and progression is driven mainly by activated PI3K/AKT pathway. Activation of this pathway is caused either by PTEN mutation which occurs in 5–20% of cases, hypermethylation of PTEN promoter [50] or to a lower extent by mutations in PI3K. Constitutive activation of PI3K/AKT signalling not only stimulates melanoma proliferation and survival but also augments its invasiveness [21], chemoresistance [6], and vascular mimicry [51]. The emerging role of PI3K/AKT signalling in acquisition of resistance to BRAF inhibitors by melanoma cells [52] resulted in development of molecules targeting PI3K/AKT. Currently, some of them are being tested as potential drugs for overcoming resistance to targeted therapy [45].
Activation of MAPK and PI3K/AKT pathways guarantees the malignant cells not only uncontrolled cell divisions but also immortality which is the hallmark of malignant transformation [53]. Similar cooperation in promotion of melanocytes transformation was observed for MAPK and WNT/β-catenin pathways. β-catenin can silence the promotor of P16INK4A leading to avoidance of senescence [54]. Although mutations in β-catenin gene are relatively rare, 23% of melanoma cell lines harbour activated β-catenin and in 30% of primary melanomas this protein accumulates in the nucleus [55]. It appears that canonical WNT pathway is responsible for melanocytes transformation and proliferation, while non-canonical WNT signalling (mediated by Wnt5A) is involved in a more invasive growth of melanoma cells. In this way, WNT pathway facilitates the capacity of melanoma cells for switching between the proliferating and invasive phenotype [56]. WNT/β-catenin pathway is also involved in immune evasion. 45% of melanomas with active β-catenin lacks infiltrating lymphocytes T which makes these tumours unresponsive to immunotherapy [57].
The aforementioned molecular changes (summarised in Table 2) guarantee development of proliferating and invasive melanoma (Figure 1). However, growth and spreading of melanoma cells would be impossible if they were recognised and effectively eliminated by the immune system.
Table 2
The main signalling pathways involved in melanoma development
| Signalling pathway (~% cases) | The main proteins and mechanism of activation/suppression | Occurrence (% of cases) | Role in melanoma development | References |
|---|---|---|---|---|
| MAPK (90%) | BRAF mutation NRAS mutation C-KIT mutation | 50–70% 20–30% 2–5% | Proliferation | [5–7] |
| PI3K/AKT (60%) | PTEN mutation PTEN methylation PI3K mutation | 5–20% 60% 3% | Proliferation, cell survival | [6, 50] |
| RB (100%) | *CDKN2A (P16) – mutation – Methylation – Expression of inhibitor ID1 – Activation of β-catenin signalling pathway – Activation of cyclin D1 oncogene | 75% | Cell cycle regulation, senescence and cell death, immortality | [6, 43, 46, 94] |
| TERT (75%) | *TERT – mutation in promotor sequence | 74% cell lines analysed 71–77% clinical samples | Inhibition of senescence and induction of immortality | [22, 23, 95, 96] |
| P53 (90%) | *CDKN2A mutation TP53 mutation MDM2, MDM4 MDM4 – overexpression | 30–70% 10% 4%, 7% 65% | Impairment of apoptosis, survival | [5, 41, 44, 47, 55] |
| Wnt/β-catenin | – Mutations – Protein activation – Protein accumulation | – Very rare – Cell lines (23%) – Metastatic melanomas (28%) | Inhibition of senescence, proliferation, invasive growth, cell plasticity, resistance to therapies, immunosuppression | [5, 55] |
| MITF | MITF amplification SOX10 mutation | 10% (primary), 21% (metastasis) 9% (primary melanomas) | Cell proliferation and survival, dedifferentiation | [34, 35] |
| NOTCH | – FBXW7 mutation – Activation of Akt pathway – Hypoxia | 8% metastasis; 40% cell lines – protein inactivation | Cell proliferation and survival, invasiveness | [25, 97] |
| STAT3 | – Activation of Src kinase | Most analysed cell lines | Proliferation, survival, angiogenesis immunosuppression, metastasis | [98, 99] |
Figure 1
Neoplastic transformation of melanocyte. Upon mutation in BRAF or NRAS gene, melanocyte acquires the ability to sustained proliferation. Subsequent mutation in the promotor of TERT enables the cell to avoid senescence while inhibition of RB and p53 pathways suppresses cell cycle control and apoptosis. At this point melanocyte turns into melanoma cell. Further activation of PI3K/AKT pathway and involvement of other tumour-related proteins (e.g. MITF, WNT, STAT3, NOTCH) regulates such processes as invasive growth, induction of angiogenesis and immunosuppression. Finally, all the aforementioned molecular changes lead to development of aggressive, metastatic melanoma cell
Immune evasion and immunotherapy in melanoma
Immune evasion is another hallmark of malignant cells. It is a strategy used by cancer cells to evade the host’s immune response [58]. There are two mechanisms of immune evasion: the impairment of lymphocytes T recruitment due to failed production of specific chemokines and inhibition of activation of these cells due to T-cell anergy, suppression by regulatory T cells, or ligation of negative regulatory receptors. The latter mechanism involves expression of cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and programmed cell death 1 (PD-1) proteins on T cells [59] and programmed death ligand-1, -2 (PD-L1, -2) on melanoma cells [60]. They are molecules involved in inhibition of anti-tumour T-cell response.
CTLA-4 (CD152) is a member of the immunoglobulin superfamily which transmits an inhibitory signal to T cells. In response to T-cell activation, CTLA-4 expression on T-cells is up-regulated to compete with binding of CD28 to B7 (CD80) ligand on antigen presenting cells (APCs). It binds to CD80 with approximately 100-fold greater affinity than the CD28 receptor. The binding causes inhibition of T-cells and subsequent immunosuppression. Consequently, CTLA-4 blockade can enhance endogenous anti-tumour immune responses as demonstrated in preclinical and clinical trials of two human anti-CTLA-4 monoclonal antibodies: ipilimumab and tremelimumab [61]. The improvement in the survival rate as compared to dacarbazine (DTIC) treatment led to the approval of ipilimumab for the treatment of unresectable metastatic melanoma. In the USA on the basis of two clinical trials [62, 63], ipilimumab was also approved for adjuvant treatment of patients after lymphadenectomy (stage III). The promising results opened the door for yet another checkpoint inhibitor, nivolumab, a human anti-PD-1 antibody.
PD-1 is a T-cell co-inhibitor receptor activated through the binding with two ligands, PD-L1 (B7-H1, CD274) and PD-L2 (CD273). The previous one is expressed on resting B cells, T cells, macrophages and DCs [64] as well as on various types of tumour cells including melanoma [65]. Its expression is associated with worse prognosis [66]. PD-L1 interaction with its receptor on T cells, PD1 (CD279), inhibits lymphocytes T activation and production of cytokines. Consequently, the PD-1/PD-L1 axis serves as a major inhibitory checkpoint for activated T cells. Activation of this signalling in normal tissues protects against autoimmunity, while in tumours it stimulates immune evasion.
The exact mechanism of PD-L1 induction on tumour cells is not known. The data suggests involvement of NF-κB [67], STAT3 [68, 69] and post-transcriptional events controlled by miR-17-5p [70]. Also cytokines such as IL-1 [71], IL-6 [72] and predominantly IFN-γ [63, 59] seem to be a strong driver of PD-L1 expression. The latter one is produced by tumour infiltrating T cells (TILs), which paradoxically may contribute to immune resistance of the tumour [73]. In response to an initial anti-tumour immune attack, tumour cells establish a microenvironment (expression of PD-L1) that promotes checkpoint inhibition. This phenomenon is known as “adaptive immune resistance”. In contrast, when PD-L1 is expressed constitutively due to genetic changes, this phenomenon is called “innate/intrinsic immune resistance”, however it does not seem to be the case in melanoma [74].
So far two drugs targeting PD-1 molecules on T cells have been approved by the FDA for advanced melanoma treatment: nivolumab [4] and pembrolizumab [75]. They are becoming drugs of first choice in melanoma immunotherapy due to their better performance in clinical trials than that of ipilimumab. They both have a similar overall response rate (ORR) and efficacy. Almost 60% of patients treated with nivolumab survived 2 years [76], while 50% of patients treated with pembrolizumab lived longer than 2 years (33 months) and 98% of them achieved long control of the disease (KEYNOTE-006 study). Even better results were achieved for a combination of nivolumab and ipilimumab [77]. Currently, many new immunotherapeutic drugs including PDL-1 (Figure 2) and other inhibitory receptors antagonists (e.g. TIM-3, LAG-3) as well as agonists of costimulatory receptors (e.g. varlilumab; Table 1 B) are being tested. This may give rise to many new combinations of various immunotherapeutic drugs, targeted drugs as well as combinations of checkpoint inhibitors with targeted drugs (e.g. atezolizumab in combination with vemurafenib plus cobimetinib).
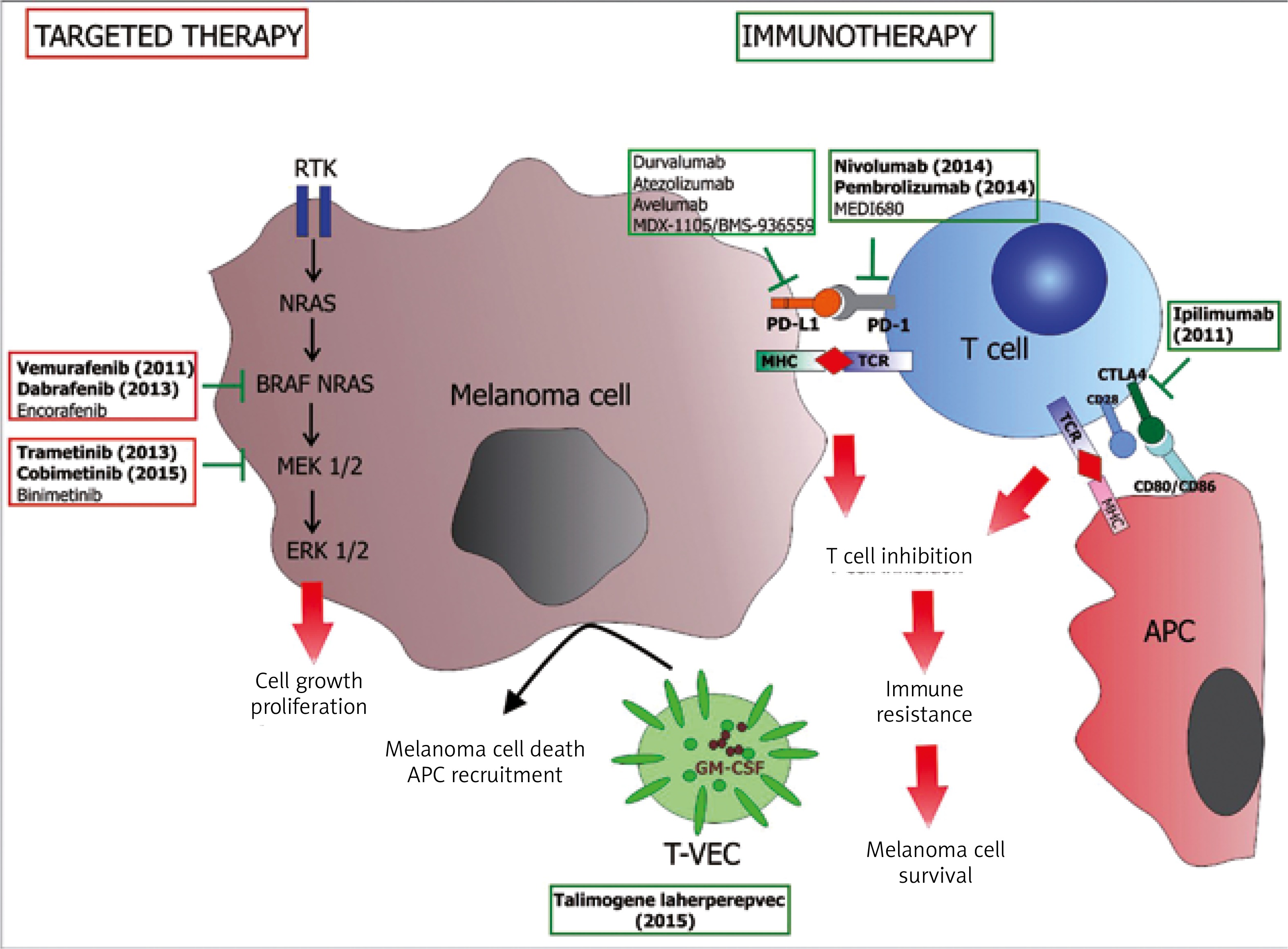
Figure 2
Novel therapies for advanced melanoma. Since 2011 eight new drugs have been approved for advanced and disseminated melanoma treatment. They constitute two main therapeutic approaches: targeted therapy and immunotherapy. Targeted drugs are synthetic inhibitors of BRAF or MEK kinases aimed at inhibition of constitutively active MAPK pathway. Four drugs of this kind were approved by the FDA as mono- or combination therapy (vemurafenib, dabrafenib, trametinib and cobimetinib) while two white two other have been approved only in combination (encorafenib and binimetinib). Immunotherapy is mainly based on treatment with antibodies against immune checkpoints on T cells such as CTLA4 (ipilimumab) and PD-1 (nivolumab and pembrolizumab). A few new drugs of this kind (including inhibitors of PD-L1) are being tested in clinical trials. Immunotherapy also involved treatment with genetically modified oncolytic virus (Talimogene laherparepvec, T-VEC) dedicated to treat regional and cutaneous metastasis
Another type of immunotherapy approved by the FDA for melanoma treatment is talimogene laherparepvec (T-VEC). It is an engineered oncolytic herpes simplex type 1 virus with neurovirulence factor replaced by granulocyte-macrophage colony-stimulating factor (GM-CSF). T-VEC is directly injected into melanoma tumours (regional or cutaneous metastasis) where it induces melanoma cell death and by local production of GM-CSF which recruits antigen presenting cells to tumour microenvironment, enhances dendritic cell function and promotes cytotoxic T-cell responses to tumour-associated antigens. In clinical trials T-VEC showed a 25% objective response. In 16% of patients the response was durable. It seems that T-VEC may induce immune response in non-injected lesions, however it does not occur frequently in visceral metastasis [78].
Future perspectives
The long-awaited progress in advanced melanoma treatment is a fact. However, due to intrinsic and acquired resistance most patients do not take advantage of it. The current challenge is not only implementation of novel combination therapies but also development of predictive tests for personalised treatment. The combination therapies will involve drugs already approved as well as the emerging ones still in clinical trials (Table 1 B). It appears that further improvement in melanoma treatment will depend not only on efficacy of the drugs but also on the combinations used, therapeutic schemes applied and most of all proper selection of patients. The latter challenge, however, cannot be met without deeper knowledge of melanoma biology and elucidation of the influence of non-tumour-related factors (e.g. microbiome, immune system) on treatment response. In the future, this knowledge together with novel diagnostic tools such as NGS (Next-Generation Sequencing) and liquid biopsy may contribute to further improvement in melanoma treatment.







