
Introduction
Hepatic encephalopathy (HE) is a clinical complication defined as an elevated serum and brain level of ammonia because of liver failure. Acute and chronic liver injury induced by diseases or xenobiotics is associated with hepatic failure, impairment of ammonia detoxification, and HE [1]. HE and hyperammonemia could lead to brain injury, cognitive dysfunction, coma, or even patients’ death [1].
Ammonia is the most suspected neurotoxic molecule involved in the pathogenesis of HE [2, 3]. Several mechanisms have been proposed for ammonia neurotoxicity [3-6]. Oxidative/nitrosative stress, inflammatory response, excitotoxicity, and disturbances in cellular calcium homeostasis are proposed to be involved in ammonia neurotoxicity [3-6]. Mitochondrial impairment and brain energy crisis represent another interesting mechanism for ammonia-induced brain injury [3, 7-11]. It has been found that ammonia is accumulated in mitochondria through specific transporters [9, 12]. Hence, this toxic molecule reaches a high level in the mitochondrial matrix [9, 12]. It has been found that NH4+ could inhibit vital enzymes involved in energy metabolism [3, 13-16]. Moreover, the effects of NH4+ on the mitochondrial electron transport process, mitochondrial membrane potential, and mitochondrial permeabilization have been well documented [3, 9, 12]. These events could lead to the collapse of mitochondrial adenosine triphosphate (ATP) production and a brain energy crisis. Moreover, NH4+-induced mitochondrial permeabilization could lead to the release of cell death mediators from this organelle [3]. NH4+-induced brain mitochondrial impairment and energy crisis could be connected to cognitive dysfunction or permanent brain injury during HE [3]. All these events highlight the pivotal role of mitochondrial impairment in the pathogenesis of HE.
N-acetyl cysteine (NAC) is used as a drug of choice against acetaminophen hepatotoxicity [17]. On the other hand, several pharmacological properties have been identified for NAC [18]. NAC has been widely investigated for different forms of xenobiotic-induced toxicity [19-22]. The positive effects of NAC supplementation against different diseases also have been documented [18, 23, 24]. The effects of NAC on oxidative stress and its associated events is the most plausible mechanism for its protective properties in biological systems [24, 25]. NAC could directly interact with reactive species [24, 25]. Moreover, NAC is able to significantly enhance cellular enzymatic antioxidant defense systems [24, 25]. NAC administration preserves cellular glutathione (GSH) at a high level [24, 25]. GSH and its associated enzymatic systems are the most pivotal cellular antioxidant mechanisms [24, 25]. Interestingly, the positive effects of NAC on mitochondrial function also have been mentioned in previous studies [26-29]. It has been found that NAC could significantly enhance cellular ATP levels and prevent mitochondria-mediated cell death and organ injury [26-29].
The effects of NAC on several neurological disorders have been investigated [23, 30]. On the other hand, previously, we found that NAC administration could significantly suppress brain oxidative stress biomarkers and improve locomotor activity in APAP-treated animals [31]. The current study was designed to evaluate the effects of NAC on brain mitochondria and energy level as pivotal mechanisms involved in ammonia neurotoxicity.
Material and methods
Chemicals
4,2-Hydroxyethyl,1-piperazine ethane sulfonic acid (HEPES), 6-hydroxy-2,5,7,8-tetramethyl chroman-2-carboxylic acid (Trolox), phenol, thiobarbituric acid (TBA), reduced GSH, rhodamine 123, 2´,7´-dichlorofluorescein diacetate (DCFH-DA), malondialdehyde (MDA), potassium chloride (KCl), sodium phosphate dibasic (Na2HPO4), ethylenediaminetetraacetic acid (EDTA), oxidized glutathione (GSSG), Coomassie brilliant blue, methanol HPLC grade, ethylene glycol-bis (2-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA), mannitol, acetonitrile HPLC grade, sucrose, and NAC were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Acetyl-para-aminophenol (APAP; acetaminophen), trichloroacetic acid (TCA), calcium chloride, hydroxymethyl aminomethane hydrochloride (Tris-HCl), and ammonium chloride were purchased from Merck (Darmstadt, Germany). All salts for preparing buffer solutions were of analytical grade and prepared from Merck (Darmstadt, Germany).
Animals
Male C57BL/6J mice (n = 40; 20-25 γ weight) were obtained from the Animal Breeding Center, Shiraz University of Medical Sciences, Shiraz, Iran. Animals were housed in a standard environment (ambient temperature of 23 ±1ºC, a 12 L : 12 D photoschedule, along with ≈40% relative humidity). Mice had free access to a normal rodents’ diet (RoyanFeed, Laboratory animals’ food company, Esfahan, Iran) and tap water. Animal handling and use were approved by the ethics committee of Shiraz University of Medical Sciences (#14822).
Animal model of fulminant hepatic failure
Acetaminophen (APAP)-induced acute liver failure was induced by a single dose of the drug (800 mg/kg, i.p.) [32]. The treatments were as follows: 1) Control (vehicle-treated); 2) APAP (800 mg/kg dissolved in warm normal saline, 25 ml/kg); 3) APAP + NAC 100 mg/kg, i.p.; 4) APAP + NAC 200 mg/kg, i.p.; 5) APAP + NAC 400 mg/kg, i.p. NAC was administered intraperitoneally two hours after APAP. The doses for NAC, the time of treatment, as well as the APAP-induced HE model, were selected based on previous studies in this field [31, 32].
Plasma and brain ammonia
Plasma and brain ammonia levels were measured with standard kits based on the absorbance photometry method of phenate-hypochlorate reaction [33]. For assessing the NH4+ level in the brain tissue, first tissue extract was prepared. For this purpose, samples (100 mg) of the forebrain (cerebral cortex) were dissected, homogenized, and deproteinized in 3 ml of ice-cooled lysis solution (trichloroacetic acid, 6%, w/v, 4°C). After centrifugation (12,000 g, 10 minutes, 4°C), the supernatant was collected and neutralized with potassium carbonate (KHCO3; 2 mol/l, pH = 7). Afterward, brain ammonia content was measured using standard kits [33].
Brain mitochondria isolation
Mice brain mitochondria were isolated based on the differential centrifugation method [34]. Animals’ brain was excised, washed, and minced in the ice-cooled mitochondria isolation buffer (75 mM mannitol, 225 mM sucrose, 0.5 mM EGTA, 2 mM HEPES, 0.1% essentially fatty acid-free bovine serum albumin, and pH = 7.4) [34-36]. Then, tissue was homogenized in the mitochondria isolation buffer (1 : 10 v : w;buffer : tissue ratio) [34, 35, 37]. The brain tissue homogenate was centrifuged (1000 g, 20 min at 4°C) to remove intact cells and nuclei. Then, the supernatant was collected and further centrifuged at 10,000 γ (4°C for 20 min) to precipitate the heavy membrane fractions (mitochondria). This step was repeated at least three times using the fresh mitochondria isolation buffer medium. Finally, the mitochondria pellet was suspended in 5 ml of incubation buffer (220 mM sucrose, 2 mM HEPES, 70 mM mannitol, and pH = 7.4) [34, 35, 37].
Mitochondrial swelling and permeabilization
Mitochondrial permeabilization was monitored by the light scattering method, as previously described [35]. Briefly, 100 µl of isolated brain mitochondria preparations (0.5 mg protein/ml) were suspended in the pre-warmed (30°C) swelling buffer (125 mM sucrose, 65 mM KCl, 10 mM HEPES, pH = 7.2) [38]. The light absorbance was monitored (λ = 540 nm) during 30 minutes of incubation (30 s intervals; constant temperature of 30°C) [35, 39]. Decreased light absorbance is consistent with an increase in mitochondrial volume [35]. The differences between the absorbance of each sample were measured and reported as maximal mitochondrial swelling amplitude [35].
Mitochondrial ATP level
Based on a previously reported procedure, mitochondrial ATP level was assessed by HPLC [40]. Briefly, 1 ml of isolated mitochondria (10 mg protein/ml) was mixed with 100 µl of ice-cooled TCA (50% w : v, 4ºC), incubated on ice (10 min, 4ºC), and centrifuged (15,000 g, 10 min, 4ºC). Afterward, the supernatant was treated with 100 µl of ice-cooled 1 M KOH solution [40]. Then, samples were centrifuged (15,000 g, 10 min, 4ºC), and 25 µl of the supernatant was injected into an HPLC system consisting of an LC-18 column (µBondapak, 4.6 × 250 mm). The mobile phase was composed of KH2PO4 (100 mM, pH = 7 adjusted with KOH), tetrabutylammonium hydroxide (1 mM), and acetonitrile (2.5% v : v). The flow rate of the mobile phase was 1 ml/min, and the UV detector was set at λ = 254 nm [40].
Mitochondrial dehydrogenases activity
Mitochondrial dehydrogenase activity was determined by the methyl tetrazolium (MTT) assay [41-43]. Briefly, a mitochondrial suspension (1 mg protein/ml) in a buffer containing 320 mM sucrose, 1 mM EDTA, and 10 mM Tris-HCl, pH = 7.4, was incubated with 40 µl of MTT (0.4% w : v) at 37°C (30 min, in the dark). Samples were centrifuged (10,000 g, 15 min), and the product of purple formazan crystals was dissolved in DMSO (1 ml) and centrifuged (15,000 g, 5 min). Then, samples absorbance at λ = 570 nm was measured with an EPOCH plate reader (BioTek Instruments, Highland Park, USA).
Mitochondrial depolarization
The mitochondrial ability to take up the cationic fluorescent dye, rhodamine 123, was used as an index of mitochondrial depolarization [35, 44, 45]. For this purpose, the mitochondrial fractions (0.5 mg protein/ml) were incubated with 10 µl of rhodamine 123 (final concentration 10 µM) in the depolarization assay buffer containing 125 mM sucrose, 65 mM KCl, 10 mM HEPES, pH = 7.2 (20 min, 37ºC). Samples were centrifuged (10,000 g, 5 min, 4ºC) and the fluorescence intensity of the supernatant was measured using a FLUOstar Omega (BMG Labtech, Germany) multifunctional microplate reader at the excitation and emission wavelengths of λ = 485 nm and λ = 525 nm,respectively [35, 46].
Lipid peroxidation in brain mitochondria
Mitochondrial formation of thiobarbituric acid reactive substances (TBARS) was measured as an index of lipid peroxidation in this organelle [35, 47]. First, isolated brain mitochondria were washed to remove sucrose. For this purpose, mitochondria samples were added to 5 ml of an ice-cold MOPS-KCl buffer (50 mM MOPS, 100 mM KCl, pH = 7.4) and centrifuged (10,000 g, 20 min, 4ºC). The pellet was re-suspended in fresh MOPS-KCl buffer. Afterward, the mitochondrial suspension (1 mg protein/ml) was mixed with twice its volume of TBARS assay reagent (trichloroacetic acid 15% w : v, thiobarbituric acid 0.375% w : v, HCl 2 N, and Trolox 0.5 mM). Then, the mixture was heated in a water bath (15 min at 100ºC) [35, 48]. After centrifugation (10,000 g, 20 min), the absorbance of the supernatant was assessed at λ = 532 nm using an EPOCH plate reader (BioTek Instruments, Highland Park, USA) [35, 42].
Mitochondrial glutathione content
The levels of reduced (GSH) and oxidized (GSSG) glutathione in isolated brain mitochondria were measured by an HPLC method [49]. The HPLC system consisted of an NH2 column (Bischoff chromatography, Leonberg, Germany, 25 cm) and a UV detector (set up at λ = 254 nm) [49]. Buffer A (water : acetate buffer; 4 : 1 v : v) and buffer B (methanol : buffer A; 4 : 1v : v) were used as mobile phases. A gradient method with a steady increase of buffer B to 95% was applied (in 25 min). The flow rate of the mobile phases was 1 ml/min [49]. Briefly, mitochondria samples (1 ml; 1 mg protein/ml) were treated with 50 µl of meta-phosphoric acid (50% w : v). Samples were mixed well, incubated on ice (15 min, 4ºC), and centrifuged (15,000 g, 15 min, 4ºC). Afterward, 300 µl of NaOH : NaHCO3 solution (2 M : 2 M) was added to the supernatant until the gas production was subsided. Then, 100 µl of iodoacetic acid (1.5% w : v in double-distilled water) was added, and samples were incubated for one hour in the dark (4ºC). Then, 2,4-dinitrofluorobenzene (DNFB; 500 µl of 1.5% w : v in absolute ethanol) was added, samples were mixed well and incubated in the dark (25ºC, 24 hours). Finally, samples were centrifuged (16,000 g, 25 min) and 25 µl of the supernatant was injected into the described HPLC system [49, 50].
Results
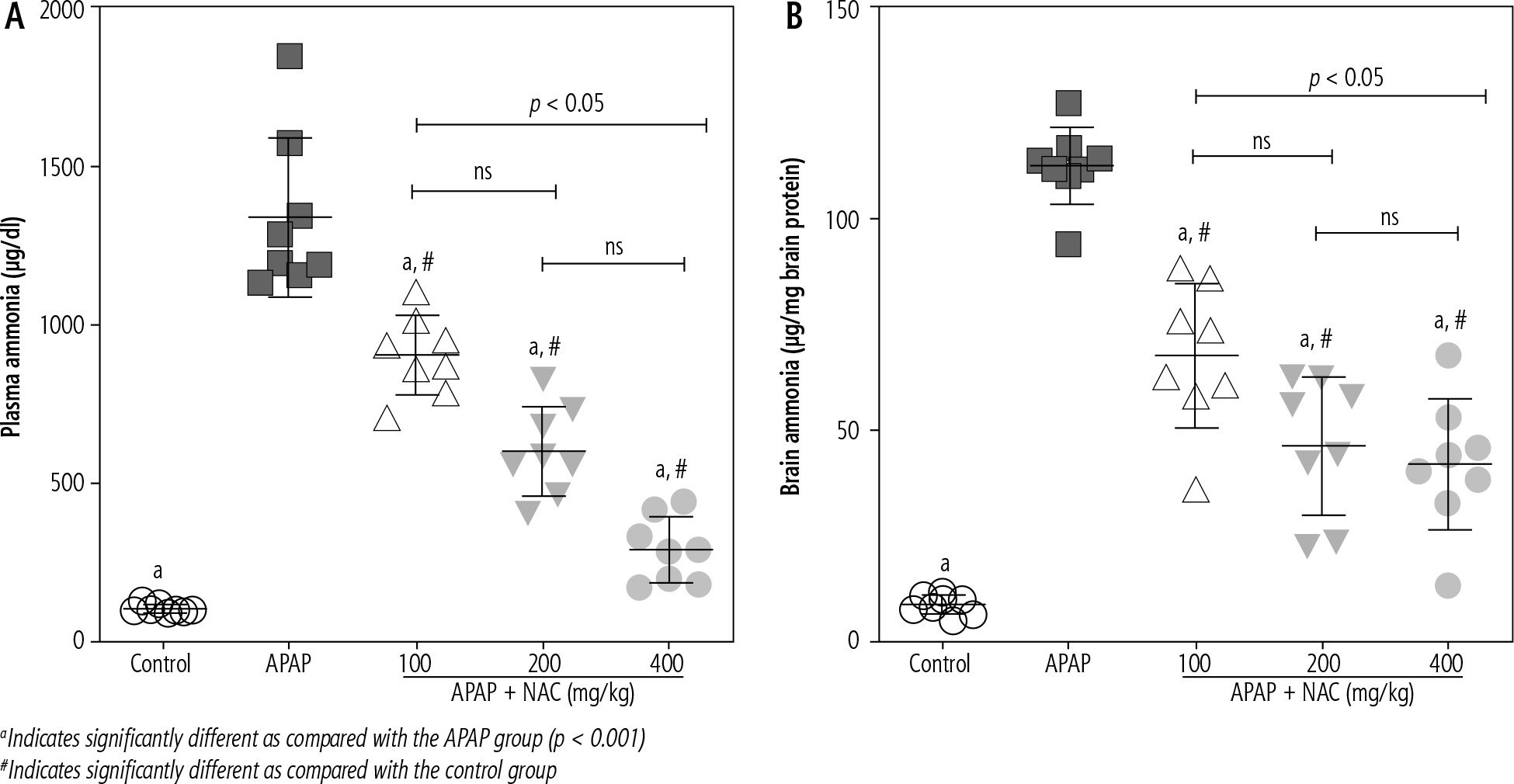
Significant elevation in plasma and brain levels of ammonium ion (NH4+) was evident in the APAP-treated animals (800 mg/kg, i.p.). On the other hand, it was found that NAC administration (100, 200, and 400 mg/kg, i.p.) significantly decreased plasma and brain ammonia levels (Fig. 1). Although the effect of NAC on plasma and brain ammonia levels was not dose-dependent in the current study (Fig. 1), the maximum effects of this compound on blood and brain ammonia was detected at the dose of 400 mg/kg (Fig. 1).
Fig. 1
Plasma and brain ammonia levels in the mouse model of acute hepatic encephalopathy. APAP – acetaminophen (single dose 800 mg/kg, i.p.), NAC – N-acetylcysteine. Data are given as mean ± SD (n = 8)
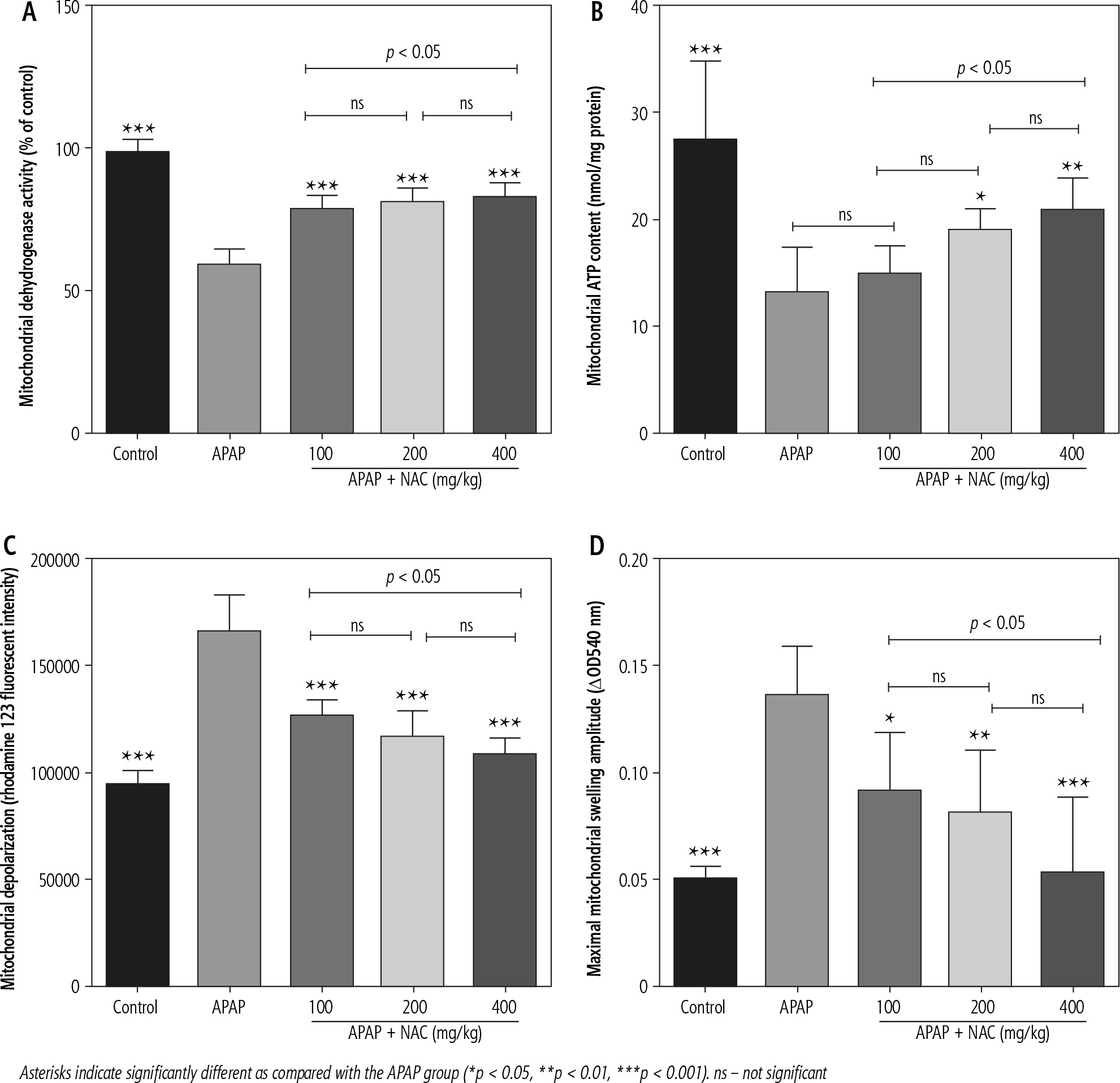
Mitochondrial indices of functionality, including mitochondrial dehydrogenase activity, ATP content, and mitochondrial membrane potential, were significantly decreased in the brain of APAP-treated (800 mg/kg, i.p.) mice (Fig. 2). Moreover, significant mitochondrial permeabilization was found in the APAP-treated group (Fig. 2). It was found that NAC treatment (100, 200, and 400 mg/kg, i.p.) improved mitochondrial function in hyperammonemic animals (Fig. 2). The effects of NAC on brain mitochondrial function were not dose-dependent (Fig. 2). However, the maximum effects of NAC on mitochondrial function were observed at the dose of 400 mg/kg of this compound (Fig. 2).
Fig. 2
Effects of N-acetyl cysteine (NAC) on brain mitochondrial indices of functionality in acetaminophen (APAP; 800 mg/kg, i.p.) model of hepatic encephalopathy. Data are given as mean ± SD (n = 8)
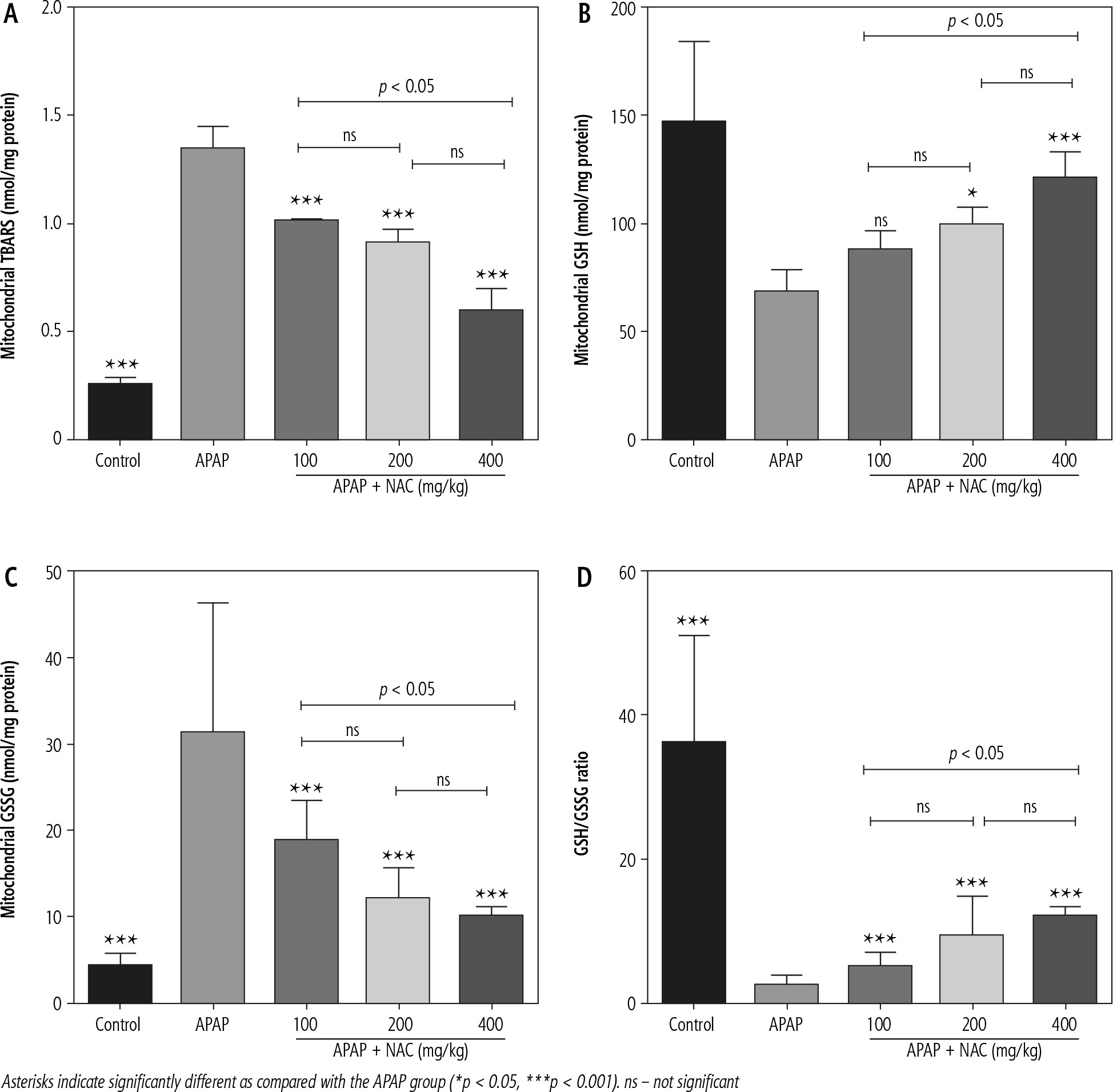
A significant increase in biomarkers of oxidative stress, including significant lipid peroxidation, increased GSSG levels, and depleted GSH content, was evident in brain mitochondria isolated from the hyperammonemic mice (Fig. 3). NAC administration (100, 200, and 400 mg/kg, i.p.) significantly mitigated biomarkers of oxidative stress in the brain mitochondria of APAP-treated animals (Fig. 3). Effects of NAC on brain mitochondria biomarkers of oxidative stress was also not dose-dependent. However, the maximum antioxidative stress effects of NAC in brain mitochondria was found at a dose of 400 mg/kg (Fig. 3).
Discussion
Hepatic encephalopathy (HE) is a serious clinical complication. The ammonium ion (NH4+) is the most suspected toxic molecule, which plays a pivotal role in the pathogenesis of HE [51-53]. NH4+ is a neurotoxic agent [51-53]. Liver failure with different etiologies leads to disturbed urea cycle and hyperammonemia [51-53]. Acute or chronic liver injury associated with xenobiotics, infectious diseases, or cholestasis/cirrhosis could induce hyperammonemia and HE [2, 54-56]. HE needs urgent and specialist treatment to prevent permanent brain injury, coma, or patients’ death.
Several interconnected mechanisms have been identified for NH4+ neurotoxicity [3, 11, 57-62]. Severe inflammation, N-methyl-D-aspartate (NMDA) receptor hyperactivation and excitotoxic response, oxidative/nitrosative stress, and mitochondrial impairment are proposed mechanisms of NH4+-induced brain injury during hyperammonemia [3, 11, 57-64]. Previously, we found that NAC administration to hyperammonemic animals significantly decreased brain tissue biomarkers of oxidative stress and improved animal locomotor activity [31]. In the current study, the effects of NAC on brain mitochondria as a crucial target of ammonia neurotoxicity were investigated.
Mitochondrial impairment and its consequent cellular energy crisis have been identified as a crucial mechanism for NH4+ neurotoxicity [3, 7, 10, 11, 57, 59, 62]. NH4+ could accumulate in the astrocytes and mitochondrial matrix through specific transporters [57, 65-67]. Afterward, NH4+ is metabolized to potentially mitotoxic metabolites such as glutamine [57]. NH4+ itself is also identified as a mitochondrial toxin [3, 57]. NH4+ or glutamine could induce mitochondrial impairment and decrease cellular energy metabolism through different mechanisms [3, 57]. It has been documented that NH4+ and glutamine are inducers of mitochondrial membrane permeability transition (mPT) [3, 57]. Induction of mPT is connected with dissipated mitochondrial membrane potential (ΔΨ), impairment of oxidative phosphorylation, and finally defective ATP production [3, 57]. MPT opening could also lead to the release of cell death mediators from this organelle [3, 57]. In the current study, significant mitochondrial permeabilization was detected in brain mitochondria isolated from hyperammonemic animals (Fig. 2).
The activity of mitochondrial electron chain complexes could be affected by NH4+ [3, 57]. On the other hand, different tricarboxylic acid cycle (TCA) enzymes, including α-ketoglutarate dehydrogenase (α-KGDH), isocitrate dehydrogenase, and malate dehydrogenase, could also be inhibited by NH4+ [3, 16, 68-70]. It has also been reported that NH4+ could significantly decrease pyruvate dehydrogenase activity [3, 14]. The inhibition of pyruvate dehydrogenase and TCA cycle enzymes will significantly decrease mitochondrial content of reducing equivalents such as nicotinamide adenine dinucleotide (NADH). NADH is essentially required for the activity of mitochondrial respiratory chain complexes. A significant decrease in the expression and activity of the mitochondrial electron transport chain (ETC) has also been documented in animal models of hyperammonemia [16, 71-75]. Inhibition of ETC by ammonia could significantly decrease energy metabolism in brain mitochondria (Fig. 2). On the other hand, NAC treatment significantly decreased mitochondrial permeability transition, improved mitochondrial membrane potential, and increased ATP levels (Fig. 2). These data could indicate the positive effects of NAC in brain mitochondria and its potential to prevent mitochondria-mediated cell death in HE (Fig. 2). On the other hand, some of the neuroprotective effects of NAC could be mediated through its indirect effects on the liver. NAC is the antidote of APAP and significantly prevents APAP hepatotoxicity. As shown in Figure 1, the level of brain and plasma ammonia is significantly decreased in NAC-treated groups (Fig. 1). These data could indicate the positive effects of NAC on the liver and enhancing NH4+ detoxification. However, the plasma and brain levels of NH4+ are still higher than those in the control group (Fig. 1). Therefore, the indirect effects of NAC (e.g., its effects on brain mitochondrial function) could also play a significant role in its protective effects during HE.
The occurrence of oxidative/nitrosative stress is a well-documented event in the brain of hyperammonemic experimental models, including the APAP model of liver failure [31, 76, 77]. On the other hand, we found that NAC administration significantly decreased oxidative stress and its associated events in the brain of APAP-treated animals [60]. Oxidative stress and mitochondrial impairment are two closely related events [78]. Cellular mitochondria are the primary sources of ROS [78]. Any defect in mitochondrial function could enhance mitochondria-facilitated ROS formation and cause severe oxidative stress [78]. Hence, mitochondrial-facilitated ROS formation could play a pivotal role in brain oxidative stress during hyperammonemia (Fig. 4) [3]. In the current study, we found that biomarkers of oxidative stress were significantly increased in the brain mitochondria isolated from hyperammonemic animals (Fig. 3). Interestingly, it has been documented that the synthesis of vital molecules such as GSH is decreased in different tissues, including the brain, during HE [79, 80]. Hence, interruption in the cytoplasmic level of GSH is a reason for lower mitochondrial GSH content (Fig. 3). On the other hand, NAC treatment also significantly decreased biomarkers of oxidative stress in the brain mitochondria isolated from the APAP-treated animals (Fig. 3). Hence, mitigation of mitochondria-mediated oxidative stress could serve as one of the pivotal mechanisms by which NAC exerts its antioxidative effect in the brain tissue during HE (Fig. 4). Indeed, many other mechanisms could also be involved in the pathogenesis of ammonia neurotoxicity [81-83]. Several studies have mentioned the role of autophagy and apoptosis in ammonia-induced astrocytes or neuron injury in different experimental models [81-83]. Evaluating the effects of therapeutic agents such as NAC on these processes might give a better insight for the clinical use of these compounds as neuroprotective agents in HE.
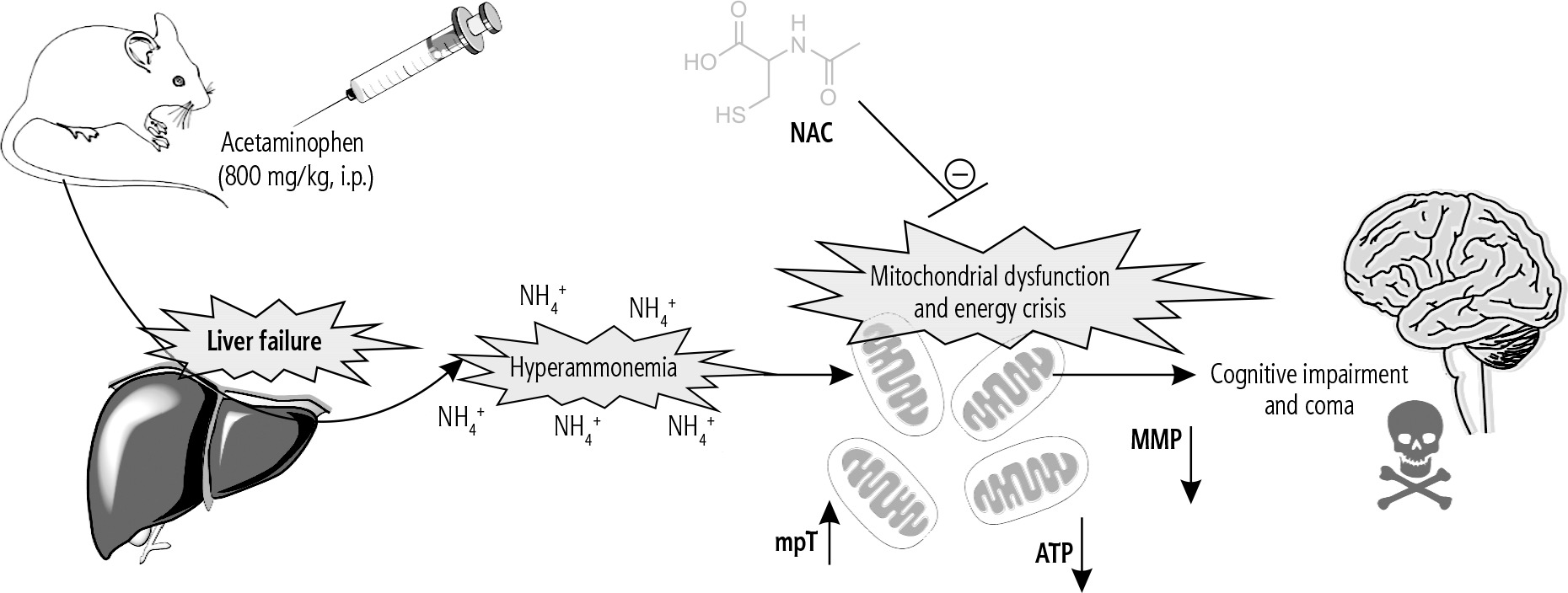
Fig. 4
The brain is the most influenced organ during liver failure and hyperammonemia. Oxidative stress and mitochondrial impairment are two interconnected events proposed to be involved in the pathogenesis of hyperammonemia-induced brain injury. In the current study, it was found that NAC administration significantly improved mitochondrial indices of functionality in the brain of hyperammonemic animals. MMP: mitochondrial membrane potential; mPT: mitochondrial permeability transition; ATP: adenosine triphosphate
Collectively, our data suggest that NAC administration not only improves brain oxidative stress biomarkers and locomotor activity in hyperammonemic mice but also significantly prevents energy metabolism disturbances in the brain. Further investigations are warranted to reveal the clinical significance of these data in patients with HE.





