
Introduction
Keratinization is the process of cytodifferentiation in which keratinocytes turn into corneocytes (cells without a nucleus) and undergo migration from the stratum basale to the stratum corneum. In this process the keratinocytes differentiate into a hardened cell, filled with protein and constituting a structurally and functionally distinct keratin-containing surface layer such as the stratum corneum. The main mass of keratin proteins consists mainly of cytokeratins and binding intermediate filament proteins (filaggrin). The main function is protection of the epidermis from mechanical damage, chemical substances (mainly acidic and alkaline substances) and ultraviolet radiation, giving the skin elasticity and maintaining the structural integrity. In humans, there are around 30 keratin families divided into two groups of acidic and basic keratins, which are arranged in pairs [1]. Normal epidermal cells contain mainly cytokeratin 1, 5, 10, 11, 14 and 15. Cytokeratin 1 and 10 (present above the epidermal basal layer) and cytokeratin 5 (found only in the basal layer) are markers of normal cell differentiation. Filaggrin, loricrin and involucrin are protein markers of terminally differentiated keratinocytes. The factor responsible for controlling the differentiation process of keratinocytes, which physiologically takes about 26–28 days (turnover time) is transforming growth factor β (TGF-β). Hyperkeratosis, parakeratosis and dyskeratosis are types of pathological keratinization.
Hyperkeratosis means excessive keratosis and thickening of the stratum corneum or disorders of proper separation of the cornified layer. Sometimes genetic defects of proteins (filaggrin, keratohyalin, keratin) and enzymes involved in cell cohesion in the horny layer, and molecules that have a critical function in the signalling pathway governing cell cohesion in the spinous layer may be responsible for this pathology.
Parakeratosis is characterized by retaining the residual nucleus in the stratum corneum and is the result of an accelerated and incomplete keratosis. Turnover time is shortened to 3–4 days.
In the spinous layer involucrin appears, a marker of terminal keratinization.
Dyskeratosis is the process of abnormal keratinization occurring prematurely within individual cells or groups of cells below the stratum granulosum.
Disorders of keratinization can be responsible not only for skin lesions but also elicit ocular changes such as blepharitis, entropion or ectropion, causing conjunctivitis, keratitis, keratopathy, corneal ulcerations, episcleritis, uveitis or even sight-threatening ocular changes like corneal perforations leading to phthisis bulbi, proliferative retinopathy, retinal vasculopathy or birdshot chorioretinopathy [2–6].
Patients with diseases involving pathological keratinization may complain about a wide spectrum of ophthalmological symptoms and report decreased visual acuity, blurred vision, diplopia, tearing, photophobia, thick yellowish discharge, red eye, red swollen lid, burning and itching, acute pain or foreign body sensation.
Ocular changes associated with hyperkeratosis
Ichthyoses
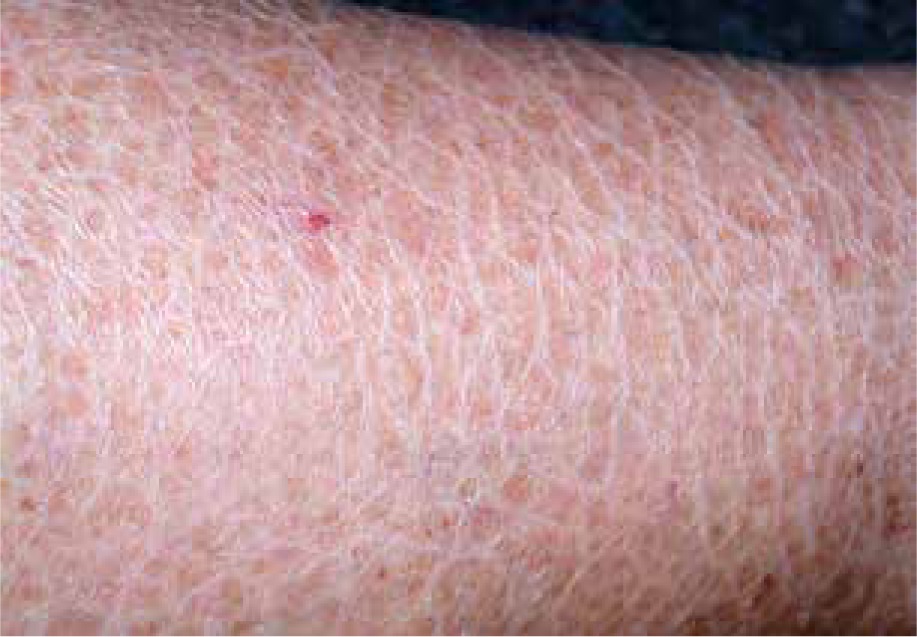
The ichthyoses apply to a heterogeneous group of disorders that can be congenital (ichthyosis vulgaris, lamellar ichthyosis, X-linked ichthyosis, non-bullous congenital ichthyosiform erythroderma) or acquired (paraneoplastic syndrome, leprosy, sarcoidosis, hypothyroidism, malabsorption syndromes, poor diet) and share as their main feature, a dry rough skin with marked scaling and without signs of inflammation [6] (Figure 1). The ichthyoses may affect the eyelids, causing hyperkeratotic lesions, madarosis, trichiasis, eyelash retraction, scales on the lashes, meibomitis, ectropion, abnormal movements of the upper eyelids, cicatricial lagophthalmos, resulting in corneal exposure and damage, which can lead to corneal perforation (Figure 2) and vision loss [7–9]. However, severe corneal damage appearing in the course of lamellar ichthyosis without ectropion has been reported, which is particularly important to a clinician to refer patients suffering from this rare disease to a routine ophthalmological examination [6]. A case of bilateral spontaneous corneal perforation with cicatricial ectropion treated with amniotic membrane transplantation with eyelid construction following minimal ectropion formation and inferior corneal leukoma seen bilaterally 6 months after the operation has been reported [6].
Figure 1
Ichthyosis vulgaris with signs of dry rough skin with marked scaling with no signs of inflammation
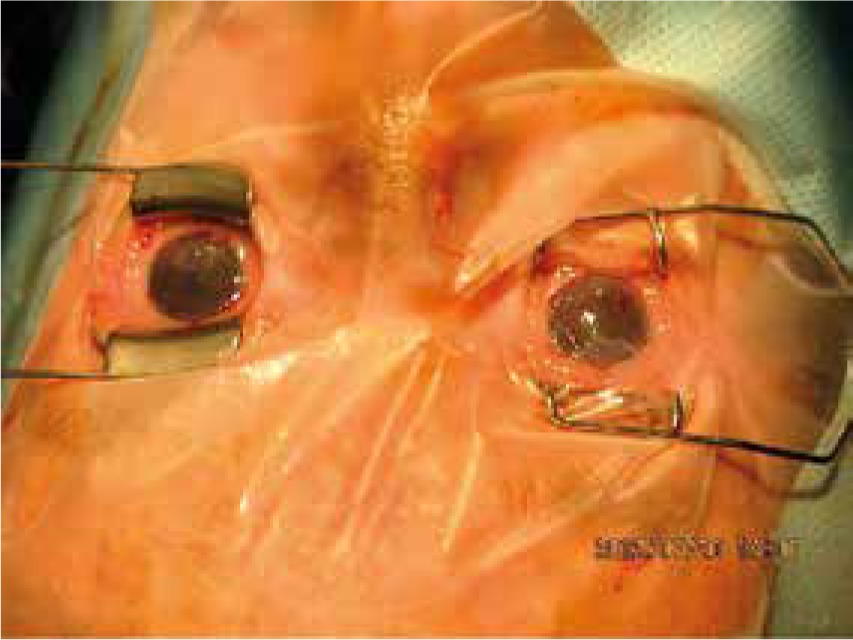
Chronic palpebral or bulbar conjunctivitis, keratinization or exposure keratopathy can occur in lamellar ichthyosis because ectropion affects the upper eyelids more than the lower eyelids [7].
The conjunctiva in ichthyoses are endangered in association with disorders of the eyelid and may become hyperaemic, thickened (evenly keratinized) due to chronic exposure and appearing chemosis [9].
Other corneal surface changes like recurrent corneal epithelial defects, punctate keratitis, keratoconjunctivitis sicca, corneal leukoma or neovascularization that may also endanger vision functions, which are observed in the congenital syndrome Keratitis–ichthyosis–deafness (KID), as hypotheses suggest, can be a result of limbal stem cell deficiency, keratinization of the meibomian glands and lacrimal ducts [8, 10]. A case of ocular surface squamous cell neoplasia in the course of KID syndrome (Keratitis–ichthyosis–deafness) has been reported [8]. Characteristic corneal stromal opacities (small punctate lesions in the deep corneal stroma) usually occur in men with X-linked ichthyosis and help in distinguishing this disease from other types of congenital ichthyoses (lamellar ichthyosis, ichthyosis vulgaris), which can be used to identify female carriers of a defected gene. They do not affect vision [9]. Corneal findings in X-linked ichthyosis include epithelial irregularity, subepithelial and anterior stromal opacities. Ophthalmic features of photophobia, corneal scars, punctate keratopathy, corneal erosion, allergic keratoconjunctivitis, superficial and deep corneal neovascularization, horizontal nystagmus and myopia may occur in ichthyosis follicularis, alopecia and photophobia syndrome (IFAP). The cornea should be monitored closely due to the risk of severe visual loss and corneal perforation in patients with ichthyosis [6]. Thinner central corneal thickness (CCT) and lower compensated corneal intraocular pressure may be present in patients with ichthyosis vulgaris that should be taken into account in refractive surgeries [9, 11]. An interesting fact is the association between ichthyosis vulgaris and atopic conditions, such as atopic dermatitis and possible corneal changes like atopic keratoconjunctivitis (allergic) which is a contributing factor to corneal ectatic disorders (keratoconus, pellucid marginal degeneration) [11]. Dry eye due to tear evaporation with decreased tear film break-up time, restriction of total closure of the eyelids and Meibomian gland dysfunction are also one of the reported ophthalmological problems in these patients [4]. Nuclear cataract or other opacities associated with the ichthyoses may exist in the lens (usually in individuals over 40 years of age) [9]. Other ocular findings related to congenital ichthyoses include coloboma of the iris, choroid and retina, optic neuropathy and crystalline macular dystrophy. A case of basal cell carcinoma of the eyelid masquerading as a chronic ectropion in the course of lamellar ichthyosis has been reported [12].
Pachyonychia congenita
Pachyonychia congenita is an inherited autosomal-dominant (the most common) or autosomal-recessive disorder of keratinization, including four types. Plantar keratodermas, nail dystrophy and hypertrophy, oral leukokeratosis, palmo-plantar hyperhidrosis and epidermal cysts are present symptoms common to all four types. Bushy eyebrows and corneal leukokeratosis can be ocular manifestations of this rare syndrome [13].
Pityriasis rubra pilaris
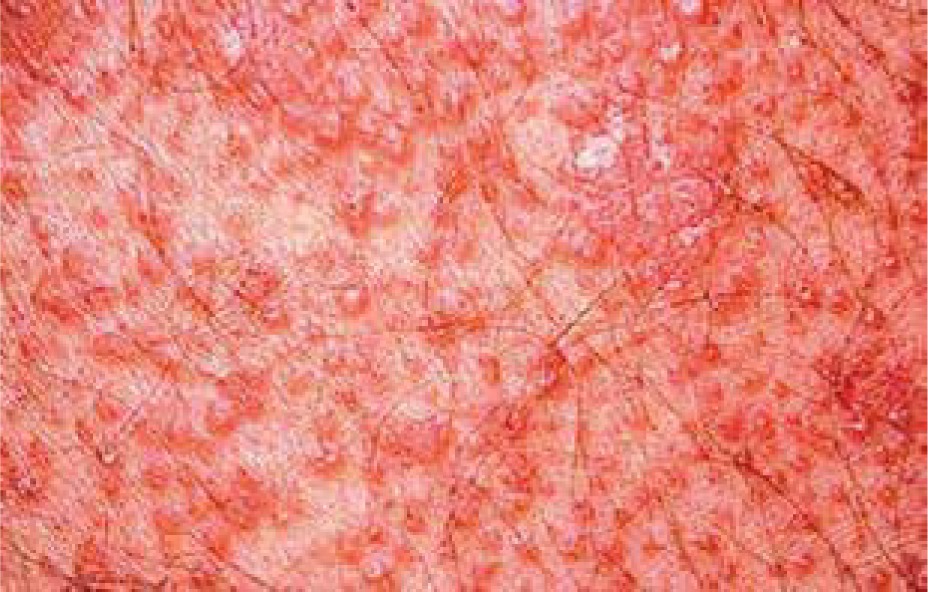
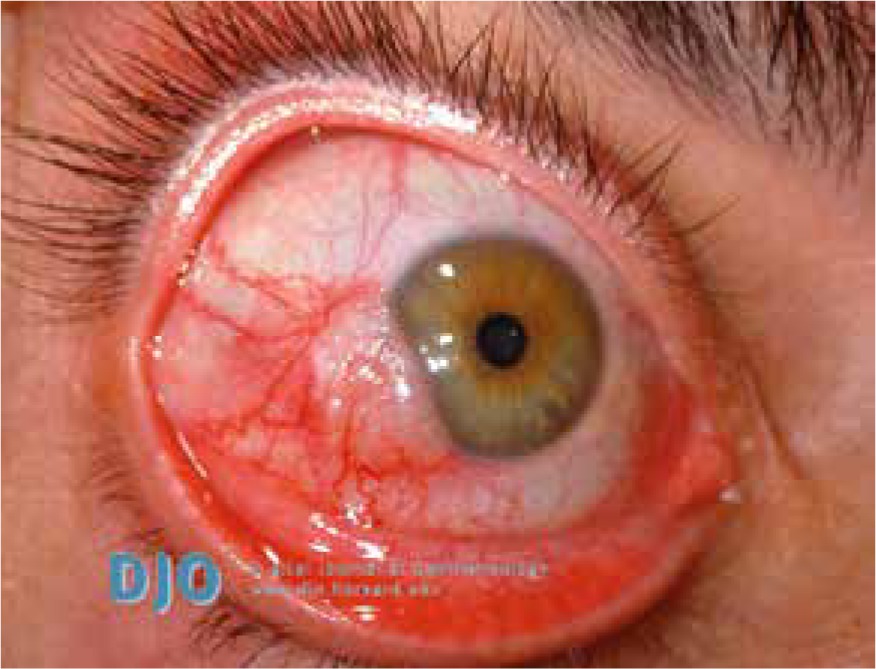
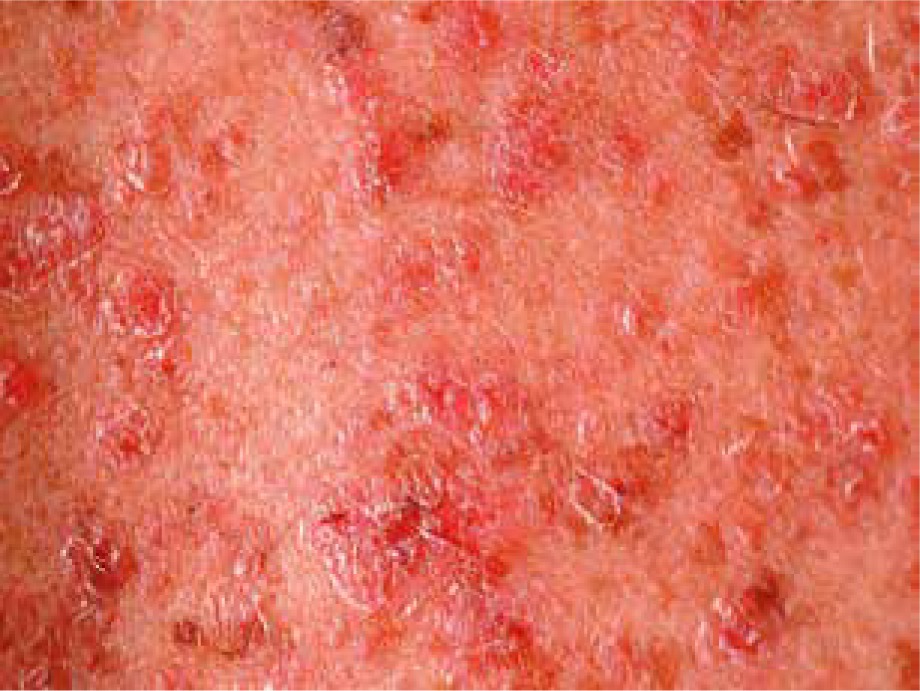
Pityriasis rubra pilaris is an idiopathic, inflammatory, papulosquamous disorder, characterized by keratotic follicular papules, reddish-orange scaly erythroderma (Figure 3), and palmo-plantar keratoderma, involving skin, nails, mucous membranes, and the eye surface. Associated ocular signs may be ectropion (also cicatricial) (Figure 4), dry eye, corneal changes including connective tissue invasion of Bowman’s zone, linear streaks, interstitial keratitis, epithelial erosions, and conjunctival keratinization, peripheral ulcerative keratitis resulting in corneal perforation [14, 15]. Pityriasis rubra pilaris may predispose patients to bilateral Herpes simplex keratitis and secondary bacterial superinfection involving a tendency of rapid stromal ulceration with the risk of perforation [15].
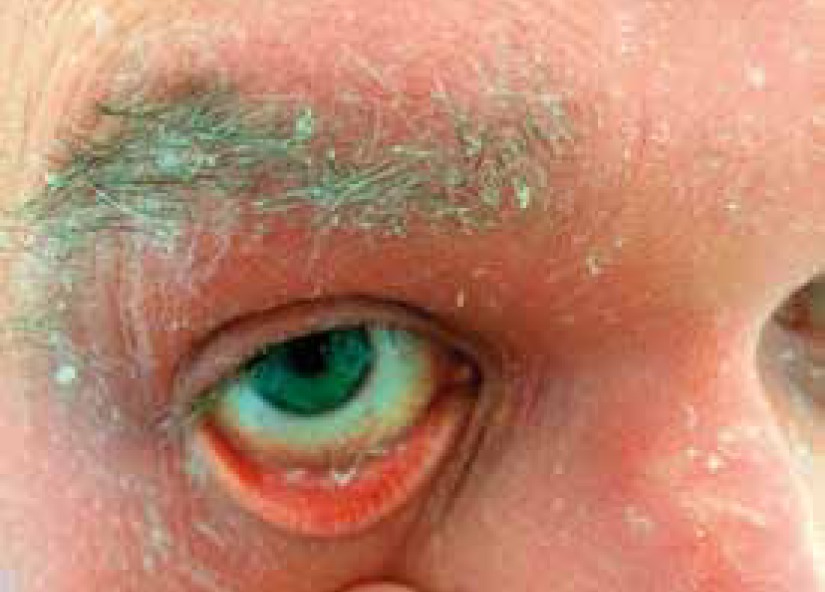
Ocular changes associated with parakeratosis
Psoriasis
Psoriasis affects 1–3% of the adult population and is related to multiple extracutaneous manifestations [16]. Ocular involvement may occur in about 10–20% of cases, is usually bilateral and oftentimes presents in the phase of acute exacerbation [17, 18]. Signs and symptoms may be subtle and easily missed during examination. Ocular involvement may occur with psoriatic plaques and is more common in men [16]. Ophthalmic complications can affect almost any part of the eye.
Relapsing orbital myositis with restriction of eye motility, mild ptosis, proptosis, conjunctival injection, symptomatic diplopia and acute orbital pain exacerbated with eye movements in the course of psoriasis with psoriatic arthritis with diplopia in a child has been reported [19].
Eye motility disorders can occur as Brown syndrome (limitation of adduction to up gaze), eyelids affection including symblepharon, blepharitis with flaky, crusted skin and scales on the eyelashes, ectropion, trichiasis, madarosis, loss of eyelid tissue, meibomian glands dysfunction (gland ducts can be plugged due to increased turnover time of epithelium with decreased supply of meibum (oily substance that prevents evaporation of tears) and dry eye symptoms. Pustules on an erythematous base can appear on the eyelid in forms of pustular psoriasis [17, 20].
Eyelid and facial psoriasis is a marker of disease severity [21].
Conjunctival lesions can be separate or extend from the eyelids, presenting as demarcated yellow-reddish plaques on the palpebrae as areas of xerotic appearance on the bulbar conjunctiva or chronic nonspecific conjunctivitis, which can lead to xerosis, symblepharon, trichiasis and subsequent corneal involvement [20].
Corneal changes in psoriasis are rare, but if present, may be sight-threatening including peripheral corneal melting, sometimes extending to perforation, corneal abscess formation, neovascularization, stromal infiltrations, superficial or deep stroma opacities and punctuate epithelial keratitis [20, 22, 23].
Corneal involvement of psoriasis has three components: thickening of the epithelium with erosions, an infiltrated zone under the Bowman’s layer with superficial vascularization and a homogeneous deep stromal opacity [20].
Dry eye is reported in 18% of patients with psoriasis [20] and is associated with L-arginine deficiency [24], which together with the amino acid L-lysine is a major component contributing to the synthesis of the antimicrobially active β-defensins induced during dry eye conditions. L-arginine is also a precursor of the moisturizer urea, which is used in the treatment of dry skin diseases and in cosmetology. Urea has also been shown to be part of the tear film.
Anterior uveitis (Figure 5) occurs in 7–20% of psoriasis cases and it is suggested that it develops more frequently in patients with arthropathic (classified as a seronegative spondyloarthropathy, related to the increased incidence of HLA B-27 positivity) or severe pustular psoriasis than among patients with other types of the disease [17, 20]. Increased aqueous flare may occur in patients with psoriasis, even if the patient did not experience a previous attack of uveitis [17]. Anterior uveitis manifests as iridocyclitis. It is important because it is able to cause serious vision complications, especially in children [17].
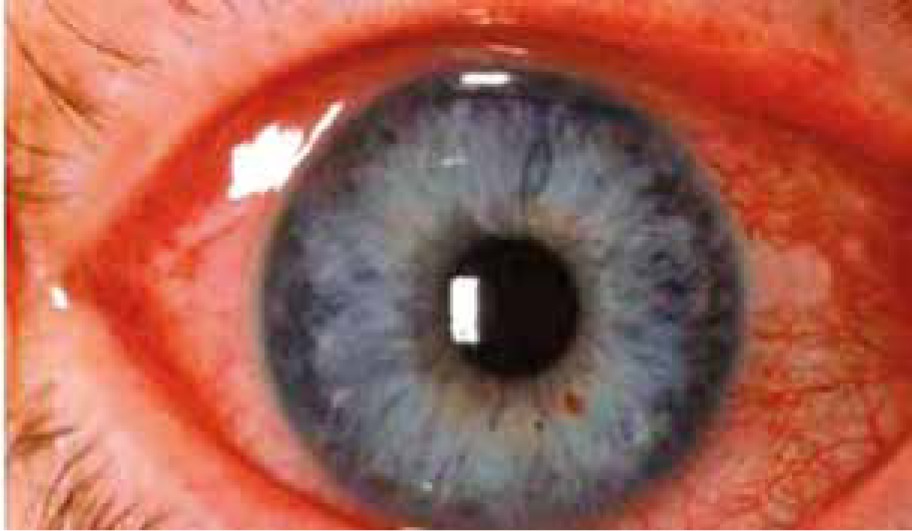
Pigment dispersion syndrome related to psoriasis has been described, which is characterized by the disruption of the iris pigment epithelium with depositions of melanin in the anterior segment, including the trabecular meshwork and possible glaucoma complications with optic nerve damage [20]. Birdshot retinopathy is the fundoscopic change which can occur in the course of psoriasis [20]. Subclinical optic neuritis has been revealed in visual evoked potential studies in psoriatic patients [25]. Retinal sensitivity may be affected by the severity of psoriasis vulgaris and is decreased due to the inflammatory response and likely due to the increased plasma levels of circulating proinflammatory cytokines [25].
Clinicians should be aware of possible ophthalmic complications, which can occur with the treatment of psoriasis and take them into account, including keratitis, conjunctival injection, dry eye syndrome, posterior subcapsular cataract, serous retinal detachment, UV keratitis, cataract or macular toxicity after methoxypsoralen use in PUVA treatment without appropriate eye protection [20, 26, 27].
A case of conjunctival squamous cell carcinoma in a patient with psoriasis treated with tumour necrosis factor-α inhibitors has been described [28].
Pityriasis lichenoides
Pityriasis lichenoides et varioliformis acuta (PLEVA) is an uncommon, acquired disease, which appears more frequently in children and juveniles. The spectrum of the disease consists of three overlapping sub-types: pityriasis lichenoides et varioliformis acuta, pityriasis lichenoides chronica, and febrile ulceronecrotic Mucha-Habermann disease (FUMHD) as a severe form of PLEVA. In severe cases of this disease spectrum, mucous membranes may be involved. Ocular involvement has been rarely reported in the form of conjunctival ulcerations and bilateral conjunctival inflammatory nodules. Patients with this disease should be provided with ophthalmological care, especially if mucous membrane changes are present [29].
Ocular changes associated with dyskeratosis
Dyskeratosis congenita
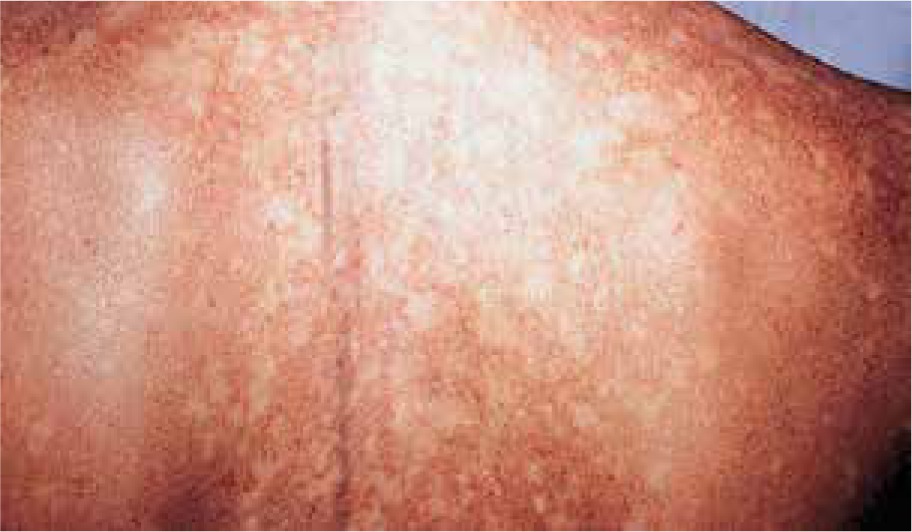
Dyskeratosis congenita is an inherited (X-linked recessive, and only occasionally autosomal dominant or recessive) multisystem disorder caused by defects in the molecular machinery responsible for telomere maintenance, which is characterized by the triad of reticular skin pigmentation (Figure 6), dystrophic nails, and oral leukoplakia. Other findings may include hyperkeratosis of the palms and feet, premature greying of hair, osteoporosis, testicular hypoplasia, pulmonary fibrosis, liver cirrhosis, increased risk of progressive bone marrow failure, pancytopenia, myelodysplastic syndrome or acute myelogenous leukaemia, squamous cell carcinoma of the head or neck and anogenital cancer [30]. Ophthalmic symptoms may occur in 30–45% of cases and may affect the eyelids with blepharitis, ectropion, entropion, trichiasis, alopecia areata of the eyelashes, pterygium, nasolacrimal duct obstruction with epiphora and conjunctivitis [3, 30, 31]. Dyskeratosis congenita is suggested to be one of the inherited causes of stem cell deficiency in the corneal limbus and that cases presenting with the stem cell deficiency in the corneal limbus might represent a subgroup of dyskeratosis congenita caused by mutations in an as yet undefined gene [31, 32]. Funduscopic changes are rare and include neovascularizations, vitreal haemorrhages, retinal haemorrhages, telangiectasia, macular exudates and oedema, retinal vasculopathy, pre-retinal fibrosis and optic nerve atrophy [3, 30, 31]. Involvement of the retinal vessels remains unclear. One hypothesis considers the presence of microembolic events in the peripheral capillaries because of dysplastic changes in the blood cells from a diseased bone marrow. The second hypothesis suggests relative hypoxia from dysfunctional macrocytic red blood cells being unable to release oxygen in the brain and retina; tissues which are particularly vulnerable to oxygen deficiency [31]. Sometimes, the main manifestation may be retinal involvement and clinicians should take an unknown retinal vasculopathy into consideration as a symptom of dyskeratosis congenita [31]. A case of bilateral proliferative retinopathy, manifested as leukocoria in a child associated with Hoyeraal-Hreidarsson syndrome (a severe form of dyskeratosis congenita) has been reported [33].
Leukoplakia
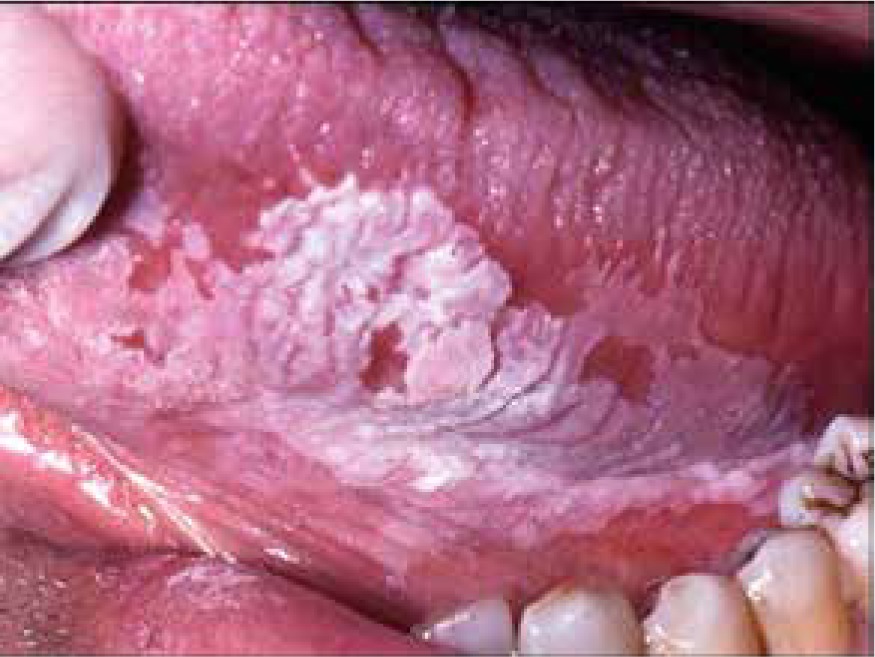
Leukoplakia is a term describing a white patch or plaque that is found mainly on the oral mucous membranes (Figure 7) which is associated with an increased risk of cancer. The aetiology remains unknown, however, the use of tobacco, alcohol abuse, irritating foods, and chronic friction, as from ill-fitting dentures or jagged teeth seem to be related to leukoplakia development on the oral mucosa [34]. Infections by the human papillomavirus (HPV) is also one suspected risk factor. Leukoplakia describes a clinical appearance, meaning a wide range of histopathological changes. It may also occur on the corneal surface, sometimes resembling a calcium deposit or mucous plaque of vernal keratoconjunctivitis. A possible reason for this may be due to chronic irritation of the cornea by the eyelashes due to entropion [34]. Leukoplakia may also appear on the limbus and in this case tends to spread over the cornea and bulbar conjunctiva [34] (Figure 8). Corneal leukoplakia may also present as so-called fingerprint epithelial lines [35].
Ocular changes associated with mixed keratinization disorders
Dyskeratosis follicularis (Darier’s disease)
Dyskeratosis follicularis (Darier’s disease) is a rare, inherited autosomally-dominant disorder characterized by abnormal epidermal differentiation and loss of normal cell-to-cell adhesion, hyperkeratosis, focal columnar parakeratosis and acanthosis due to a defective calcium transport process, encoded in the ATP2A2 gene, responsible for the calcium-dependent ATPase on the membrane of the sarco- and endoplasmic reticulum. This results in an excess of calcium ions within the sarco- and endoplasmic reticulum. The first symptoms usually appear in the mid-teens, sometimes after over-exposure to sunlight. Skin lesions include the presence of greasy, hyperkeratotic papules on middle chest, upper shoulders, neck and face (Figure 9). Ocular changes occur rarely, including dry eye, punctuate corneal epithelial defects, opacities in the periphery of the cornea, corneal subepithelial infiltrations, corneal ulcerations leading to perforations and conjunctival keratosis [36, 37]. Patients suffering from dyskeratosis follicularis are also prone to recurrent herpes keratitis and episcleritis [38]. The cornea is poorly protected due to impaired desmosome function leading to poor cell-to-cell adhesion in the corneal epithelium and related to this disorder resulting in a dry eye allowing viral colonization [38]. Involvement of the eyelids, eyelid lesions resembling basal cell carcinomas, horn-like growths along the lid margin, retinal detachment, association with retinitis pigmentosa, subnormal dark adaption and post-cataract extraction staphylococcal endophthalmitis has all been reported [36–39].
Discussion
The influence of skin diseases with pathological keratinization processes on eye and vision function is oftentimes underestimated by clinicians, which may have dramatic consequences for the patient, including vision loss due to eye surface changes (corneal ulcer and perforations), fundoscopic manifestations (retinal neovascularization, retinal vasculopathy, optic nerve atrophy) or uveitis. In severe cases such as phthisis bulbi, enucleation with insertion of a prosthesis is needed. Some patients may require laser therapy or ophthalmological operations like vitrectomy, corneal transplantations, cataract or oculoplastic surgery to save vision functions. One should be aware that the prescribed therapy may also affect the eye and its functions, causing complications, so regular ophthalmological control visits are needed for physicians and patients [20, 26–28].
The eye lesions may sometimes get expressed later than skin manifestations, such as in the case of Keratitis–ichthyosis–deafness syndrome, in which in almost 80% of cases bilateral, asymmetrical corneal vascularization is common. Knowing this fact can help saving visual functions and prevent serious complications [9]. Sometimes, the recognition of ocular changes can be helpful in setting a proper diagnosis, like in cases of corneal stroma changes, which allows to distinguish between men with X-linked ichthyosis and other types of ichthyoses and identify female carriers of the defected gene [9].
Clinicians should be aware of the fact that the most significant, early and severe clinical manifestation leading to the diagnosis may include ocular involvement, such as in cases of retinal vasculopathy and dyskeratosis congenita [31].
All physicians should have oncological alertness and remember that neoplastic processes may masquerade ocular changes, which appear in the course of diseases associated with pathological keratinization [12].
Dermatologists should regularly monitor their patients for ocular symptoms and make them vigilant, so that in a situation of the deterioration of vision, redness of the eye, acute pain, photophobia, appearance of discharge, the patients should immediately visit an ophthalmological ambulatory department. Ophthalmologists should know possible ocular changes in the course of skin diseases and take them into account when evaluating eye changes. Multidisciplinary cooperation is required to set a better prognosis and quality of life for patients.







