
INTRODUCTION
Lafora body disease (LBD) is a rare, autosomal recessive progressive myoclonic epilepsy (PME) characterized by the accumulation of aberrant glycogen-like polyglucosan inclusions, known as Lafora bodies, in neurons and peripheral tissues, including the liver, muscle, and sweat glands [1, 2]. Mutations in the EPM2A (laforin) or EPM2B/NHLRC1 (malin) genes impair glycogen metabolism, resulting in poorly branched, hyperphosphorylated glycogen precipitates that drive neurodegeneration [1, 3].
Clinically, LBD presents during adolescence, with intractable myoclonic and generalized seizures, cognitive and behavioral decline, ataxia, and visual disturbances, progressing to severe dementia and death within a decade of onset [2, 3]. The disease course is relentless, with limited effectiveness of conventional antiepileptic drugs (AEDs). Importantly, several commonly used AEDs – such as carbamazepine, phenytoin, and gabapentin – can aggravate myoclonus or worsen neuropsychiatric outcomes due to shared mechanistic or pharmacokinetic properties [4-6].
Polypharmacy, often employed in refractory epilepsy, further complicates the clinical course in LBD. Enzyme- inducing AEDs (e.g., carbamazepine, phenytoin, phenobarbital) interact significantly with co-administered agents, altering serum levels and enhancing neurotoxicity [7, 8]. Sodium valproate, although non-inducing, may inhibit hepatic metabolism and potentiate the toxicity of drugs such as phenobarbital and lamotrigine [7]. Such combinations can contribute to drug resistance, enhanced oxidative stress, and accelerated disease progression in PME patients [4, 6].
In this case report we describe a patient with biopsy- confirmed LBD who experienced rapid clinical deterio-ration despite multidrug antiepileptic therapy. The case highlights the pharmacological pitfalls of polypharmacy, underscores the need for syndrome-specific AED selection, and presents future directions for rational drug use and potential disease-modifying approaches.
Case description
An 18-year-old male presented with a four-year history of progressive neurological decline. The initial symptoms began at the age of 14, when the patient started experiencing recurrent frontal headaches, followed shortly by the appearance of involuntary, shock-like myoclonic jerks primarily involving the upper limbs. Within a few months he developed generalized tonic-clonic seizures (GTCS), which increased in frequency over time and were later accompanied by drop attacks.
As the disease progressed, the patient exhibited signi-ficant cognitive and behavioral deterioration. He became increasingly irritable and emotionally labile, withdrew socially, and began having difficulty performing daily tasks. Caregivers noted marked memory impairment, disorientation, and poor academic performance. By age 16, his symptoms had further worsened, with the deve-lopment of dysarthria, gait ataxia, and bilateral cerebellar signs. Blurred vision was also reported, although no definitive signs of cortical blindness were confirmed on examination.
Neurological assessment revealed generalized myo-clonic jerks, ataxic gait, past-pointing on finger-nose testing, and intention tremors. A Mini-Mental State Exa-mination (MMSE) score of 14/30 indicated moderate cognitive impairment. There was no history of consanguinity or similar illness in family members, and perinatal history was unremarkable.
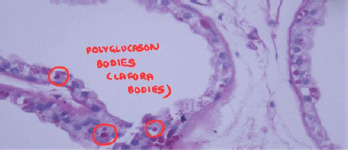
Electroencephalography (EEG) demonstrated gene-ralized polyspike-and-wave discharges at a frequency of 3-4 Hz, which were exacerbated by photic stimulation. Magnetic resonance imaging (MRI) of the brain revealed both cerebral and cerebellar atrophy. A skin biopsy obtained from the axillary region showed periodic acid- Schiff (PAS)-positive Lafora bodies within the eccrine ducts, confirming the diagnosis of LBD, a severe form of PME (Figure I).
Figure I
PAS-stained section showing intracytoplasmic Lafora bodies (polyglucosan inclusions) in eccrine duct epithelial cells (red circles), pathognomonic of Lafora body disease
While skin biopsy was historically the diagnostic standard, the current gold standard for confirming LBD is molecular genetic testing, which identifies pathogenic variants in the EPM2A (laforin) or EPM2B/NHLRC1 (malin) genes. However, in our case biopsy was pursued due to limited access to genetic testing, and diagnosis was supported by the classical clinical presentation, EEG abnormalities, and MRI findings. The routine metabolic workup, including serum lactate, ammonia, electrolytes, and liver and renal function tests, was within normal limits.
Pharmacological management
The patient was initiated on multiple AEDs in an attempt to control his worsening episodes of seizure and myoclonus. However, the regimen reflected inappropriate polypharmacy, with overlapping toxicities, pharmacodynamic redundancies, and significant pharmacokinetic interactions – which are particularly detrimental in a form of progressive myoclonic epilepsy like LBD.
The first-line treatment included sodium valproate 500 mg twice daily, a broad-spectrum AED with known efficacy in myoclonic and generalized seizures. How-ever, its concurrent use with carbamazepine 200 mg twice daily – a narrow-spectrum sodium channel blocker – was clinically counterproductive. Carbamazepine not only induced hepatic microsomal enzymes, reducing the plasma concentration and efficacy of valproate, but also worsened myoclonus and cognitive functions through neuronal excitotoxicity and oxidative stress.
The patient was also prescribed clobazam 10 mg at bedtime, a benzodiazepine that enhances GABAergic activity. While it was effective in reducing myoclonic jerks, it contributed to excessive sedation and cognitive dulling when used with other central nervous system depressants. Phenobarbitone 60 mg at bedtime, introduced for seizure control, similarly aggravated neurocognitive decline and was later discontinued. Phenytoin 100 mg twice daily, another sodium channel blocker with cerebellar toxicity, further impaired motor coordination and was also withdrawn after clinical worsening.
In subsequent months, the patient was started on levetiracetam 750 mg twice daily, an SV2A-binding AED with minimal hepatic metabolism and fewer drug interactions. It offered moderate improvement in seizure control, though mood swings and irritability were noted. Perampanel, an AMPA receptor antagonist, was introduced in a dose of 4mg twice daily for its antimyoclonic and broad-spectrum efficacy, though side effects included agitation and behavioral dysregulation. Topiramate 25 mg at bedtime, a mixed-action AED with GABAergic enhancement and glutamate antagonism, was added for additional seizure control. However, it caused cognitive dulling and weight loss.
Neuroprotective adjuncts included pyridoxine (vitamin B6) 100 mg twice daily, folic acid 5 mg once daily, magnesium, and calcium with vitamin D3. These were intended to reduce excitotoxicity and support neuronal health, though their clinical efficacy was limited in this case.
Multiple pharmacokinetic interactions were evident in this regimen: carbamazepine induced hepatic enzymes (CYP3A4), accelerating the metabolism of valproate and topiramate, reducing their therapeutic effect. Valproate, an enzyme inhibitor, increased the serum levels of phenobarbitone, risking toxicity. Overlapping sedative effects of clobazam, phenobarbitone, phenytoin, and topiramate worsened cognitive impairment and psychomotor slowing.
At the most severe stage of his illness, the patient was concurrently receiving six antiepileptic drugs – valproate, carbamazepine, clobazam, phenytoin, phenobarbitone, and topiramate (Table 1). This extreme polypharmacy contributed to excessive sedation, cognitive dysfunction, and poor seizure control, rather than additive benefit. Notably, topiramate was prescribed at a low dose of 25 mg at bedtime, well below the therapeutic range. While likely ineffective for seizure control at this dose, it still contri-buted to cognitive dulling and the suppression of appetite. This scenario exemplifies the dangers of layered AED regimens in progressive myoclonic epilepsies, where additive toxicity may outweigh therapeutic gain.
Table 1
Summary of antiepileptic drug (AED) trials, interactions, and clinical rationale
Recognizing these adverse outcomes, the clinical team revised the treatment. Drugs known to exacerbate myo-clonus or cognitive symptoms – carbamazepine, phenytoin, and phenobarbitone – were systematically withdrawn. Valproate was retained at a lowered dose of 250 mg twice daily. Levetiracetam and clobazam were continued for their relative safety in PME. Perampanel was maintained with psychiatric monitoring. The revised regimen led to reduced seizure frequency, better alertness, and improved tolerability, emphasizing the need for pharmacological tailoring over aggressive polytherapy in LBD.
A detailed summary of the pharmacological course is presented in Table 1 to highlight the rationale, interactions, and outcomes associated with each antiepileptic drug trial.
Discussion
LBD is a rare but fatal PME, distinguished by its hallmark pathological feature – the intracellular accumulation of poorly branched, insoluble glycogen aggregates known as Lafora bodies. These structures disrupt normal neuronal function and initiate a cascade of neurodegeneration marked by refractory seizures, cognitive decline, and motor dysfunction [1, 2]. Recent studies have expanded our understanding of LBD beyond neuronal pathology, revealing a critical role for astrocytes in disease progression. Astrocytes are now recognized not only as passive bystanders but also as active contributors to Lafora body accumulation, neuroinflammation, and synaptic dysfunction. Experimental models have demonstrated that targeting astrocytic glycogen synthase effectively reduces the formation of Lafora bodies and prevents neuro-degeneration, underscoring a promising glial-targeted therapeutic avenue [9].
Management of LBD remains symptomatic, with AEDs forming the cornerstone of seizure control. However, conventional polypharmacy in such cases often leads to adverse pharmacological outcomes. The patient in this case was prescribed multiple AEDs with overlapping mechanisms – particularly sodium channel blockers like phenytoin and carbamazepine – despite established evidence that such agents can worsen myoclonus, exa-cerbate cognitive deficits, and promote cerebellar dysfunction [3, 4]. These drugs, though effective in genera-lized epilepsy, are contraindicated or used cautiously in PME syndromes due to their potential to aggravate symptoms through excitatory neurotransmission imbalance and mitochondrial toxicity.
Additionally, the concurrent use of sodium valpro-ate and carbamazepine highlighted a classic phar-ma-cokine-tic and pharmacodynamic pitfall. Valproate, a broad-spectrum AED, inhibits hepatic enzymes such as CYP2C9 and epoxide hydrolase, increasing levels of concurrently administered drugs such as phenobarbitone and carbama-zepine epoxide [7]. Meanwhile, carbamazepine induces hepatic enzymes (CYP3A4), reducing the serum concentration of valproate and other AEDs. This bidirectional interaction not only diminished therapeutic efficacy but also resulted in amplified toxicity, contributing to the patient’s accelerated cognitive and neurological decline [7].
The adverse cognitive effects and psychomotor slowing observed with topiramate, clobazam, and phenobarbitone further underscore the importance of rational prescribing in complex epilepsies. Following a thorough pharmacological review, the withdrawal of these offending agents and continuation of levetiracetam, clobazam, and perampanel – with supportive neuroprotective therapy – improved the patient’s symptom trajectory and reduced sedative burden. This case reinforces the critical need for pharmacogenomic guidance and individualized treatment plans in LBD [5, 7].
While this case report is inherently qualitative, the clinical trajectory was corroborated by detailed caregiver reports and follow-up assessments. Accelerated decline was evident through worsening myoclonus, drop attacks, disorientation, and MMSE deterioration (14/30). Following AED de-escalation, caregivers reported a notable reduction in the frequency of seizures (from multiple daily drop attacks to 1-2 per day), improved wakefulness, and resumption of basic tasks like ambulation and feeding. Though formal seizure diaries or neurocognitive batteries were not employed, these real-world improvements were consistent and sustained over a three-month follow- up period. The absence of structured longitudinal tools remains a limitation but does not diminish the observable clinical benefit of pharmacological tailoring.
Emerging therapies and future perspectives
While current treatments remain symptomatic, pro-mising advances in LBD research are targeting the core pathophysiology of glycogen misprocessing. One notable intervention involves metformin, an AMPK activator traditionally used in type 2 diabetes. In preclinical murine models of LBD, metformin significantly reduced the formation of Lafora bodies, improved motor function, and preserved cognitive abilities by modulating glycogen synthesis and mitochondrial energy homeostasis [10, 11]. These findings suggest that early initiation of metformin could attenuate disease progression and improve neurological outcomes [10]. Recent attention has also turned toward cannabidiol (CBD), a non-psychoactive cannabinoid with broad-spectrum antiseizure activity. Although primarily studied in Dravet and Lennox-Gastaut syndromes, CBD has been used experimentally in LBD with mixed results. In a case report by Maini et al. [12], adjunctive CBD oil appeared to provide partial seizure relief in a patient with LBD, though cognitive and behavioral decline continued unabated. Similarly, Aso et al. [13] reviewed anecdotal use of cannabinoids in various refractory epilepsies, including LBD, noting uncertain efficacy and a need for formal trials. Despite a favorable safety profile, the utility of CBD in LBD remains preliminary, with no conclusive data on disease modification or long-term benefit. As such, its use should be considered experimental, and tailored on a case-by-case basis [12, 13]. Other experimental avenues include antisense oligonucleotides (ASOs), gene-editing tools, and glycogen synthase inhibitors, offering future hope for disease-modifying strategies [2]. One such promising ASO-based strategy involves targeting glycogen synthase (GYS1), the enzyme responsible for glycogen elongation. Ahonen et al. [14] developed a Gys1-specific ASO (Gys1-ASO), which significantly reduced brain glycogen content, prevented Lafora body formation, and mitigated neuroinflammation and seizures in mouse models of LBD. Treated animals demonstrated better survival and motor coordination without adverse effects, offering a compelling preclinical rationale for RNA-based therapeutic intervention in LBD [13]. Additionally, repurposing sodium-glucose cotransporter-2 (SGLT2) inhibitors such as empagliflozin has emerged as a novel strategy, with animal models showing reduced polyglucosan storage and improved neuroinflammatory profiles [15]. Enzyme replacement therapies (e.g., VAL-1221), while still investigational, are under clinical evaluation [16]. These rapid advancements shift the landscape toward disease-modifying therapies and reinforce the need for early molecular diagnosis to enable timely intervention.
Conclusions
This case highlights how inappropriate polypharmacy and unrecognized drug–disease interactions can worsen outcomes in LBD. While diagnosis was supported by cli-nical, radiological, EEG, and histopathological findings, the absence of molecular genetic confirmation remains a key limitation. A precision-guided pharmacological approach, coupled with access to emerging diagnostics and therapies, is essential for improving care in this rare but devastating condition.






