
Prurigo pigmentosa (PP) is a rare skin condition described first by Nagashima in 1971 [1]. Since then 259 cases have been described outside Japan, of which only 21 in Europe (Spain, Germany, Italy, Austria, Sweden). Usually PP is diagnosed in young people, mostly in their second and third decade of life [2]; the majority of patients are female; it affects women 4 to 7 times more frequently than men [3]. The cause of PP is unknown; one of the theories suggests a connection between the disease and ketosis during a restrictive diet and diabetes [3, 4].
The skin lesions are reticular and located symmetrically on the torso (usually the chest, the upper back, the lumbosacral region) and often also on the neck, the abdomen, the arms and the forehead. The evolution of the lesions, both morphological and histopathological, is characteristic for PP. In the initial stage lasting 2–3 days, intensively itching erythematous and oedematous papules appear. Within a week the lesions undergo regression and leave fuscous and brown patchy hyperpigmentation which remains on the skin up to several months. Significantly, the lesions in various stages of development can occur concomitantly [2]. In the histopathological image, the papillary dermal oedema with hyperkeratosis (orthokeratotic), necrotic keratinocytes (few), neutrophilic infiltrates (sparse and perivascular) and spongiosis (mild) are initially visible.
Fully developed, intensively itching, swollen papules are histologically visible as extravasation of erythrocytes, hyperkeratosis (orthokeratotic), lymphocytic infiltrates (patchy and lichenoid), necrotic keratinocytes (numerous) and spongiosis (moderate). The final stage of hyperpigmentation is dominated by hyperkeratosis (parakeratotic), neutrophilic infiltrates (sparse and perivascular) and melanophages in papillary dermis [2].
The patients most frequently seek consultation when the lesions are in an advanced stage of development; therefore, during the differential diagnosis, reticulate pigmentary disorders which become apparent in adults in good general health should be taken into account, such as confluent and reticulated papillomatosis (Gougerot-Carteaud syndrome), erythema ab igne, atopic dirty neck, Dowling-Degos disease, Galli-Galli disease, postinflammatory and drug-induced dermatoses [5].
Oral antibiotics, such as minocycline [6], doxycycline [7] and macrolides [8] as well as dapsone [2] are successfully used during treatment; however, postinflammatory hyperpigmentation may persist for many years following the remission of active lesions.
A 22-year-old woman was admitted to the Department of Dermatology at the Military Institute of Medicine due to recurrent skin lesions which persisted for approximately 8 years and were located on the chest, the lower back and the nape of the neck, with concurrent burning sensation and pruritus. The patient was in good general health and did not receive any medication. Prior to the hospitalization, the patient was examined by dermatologists on numerous occasions, but the test results were inconclusive – a biopsy showed non-specific lesions (with moderate oedema and scant chronic perivascular inflammatory infiltrate in the dermis; staining with CD117 did not show any mast cells), the assay of anti-mitochondrial antibodies (AMA) and the rheumatoid factor (RF) were negative – general antihistamine treatment and topical treatment with glucocorticoids were recommended, but unsuccessful.
The condition gradually progressed with periods of aggravation and minor local remissions. Upon admission, reticular, fuscous and red, erythematous and papular lesions were observed, located on the torso, in the axillary fossae, on the nape of the neck and in the left cubital fossa (Figures 1 A, B). The lesions were the most pronounced in the lumbar, subcostal and epigastric regions. In the dermoscopic image, consecutive stages of the disorder were visible. The acute stage is characterised by pink, oval lesions with superficial dotted vessels. Under pressure, those lesions show yellowish colour in the centre. The structures correspond to oedematous papules with extravasated erythrocytes. The intermediate stage is characterised by erythematous lesions, irregular in shape (without visible blood vessels) connected to one another. The regression stage is characterised by numerous grey spots visible on residual erythema. The regression image resembles the regression in lichen planus (Figures 1 C–E). The patient described the intensity of the pruritus as 10 out of 10 on the visual analogue scale (VAS). During hospitalization, the assay of antinuclear antibodies (ANA) was negative and the level of tryptase was normal. Basic laboratory tests did not show any clinically relevant deviations.
Figure 1
Photographs of the patient before treatment: fuscous and red, erythematous and papular reticular lesions, A – back, B – front, C–E – dermoscopic image: C – acute stage – pink, oval lesions with superficial dotted vessels. Under pressure, those lesions show yellowish colour in the centre; D – intermediate stage – erythematous lesions, irregular in shape (without visible blood vessels) connected to one another; E – regression stage – numerous grey spots visible on residual erythema. The regression image resembles the regression in lichen planus. F, G – Histological image: acanthosis, parakeratosis and dyskeratosis in the upper part of the epidermis, with a mixed perivascular infiltrate in the dermis
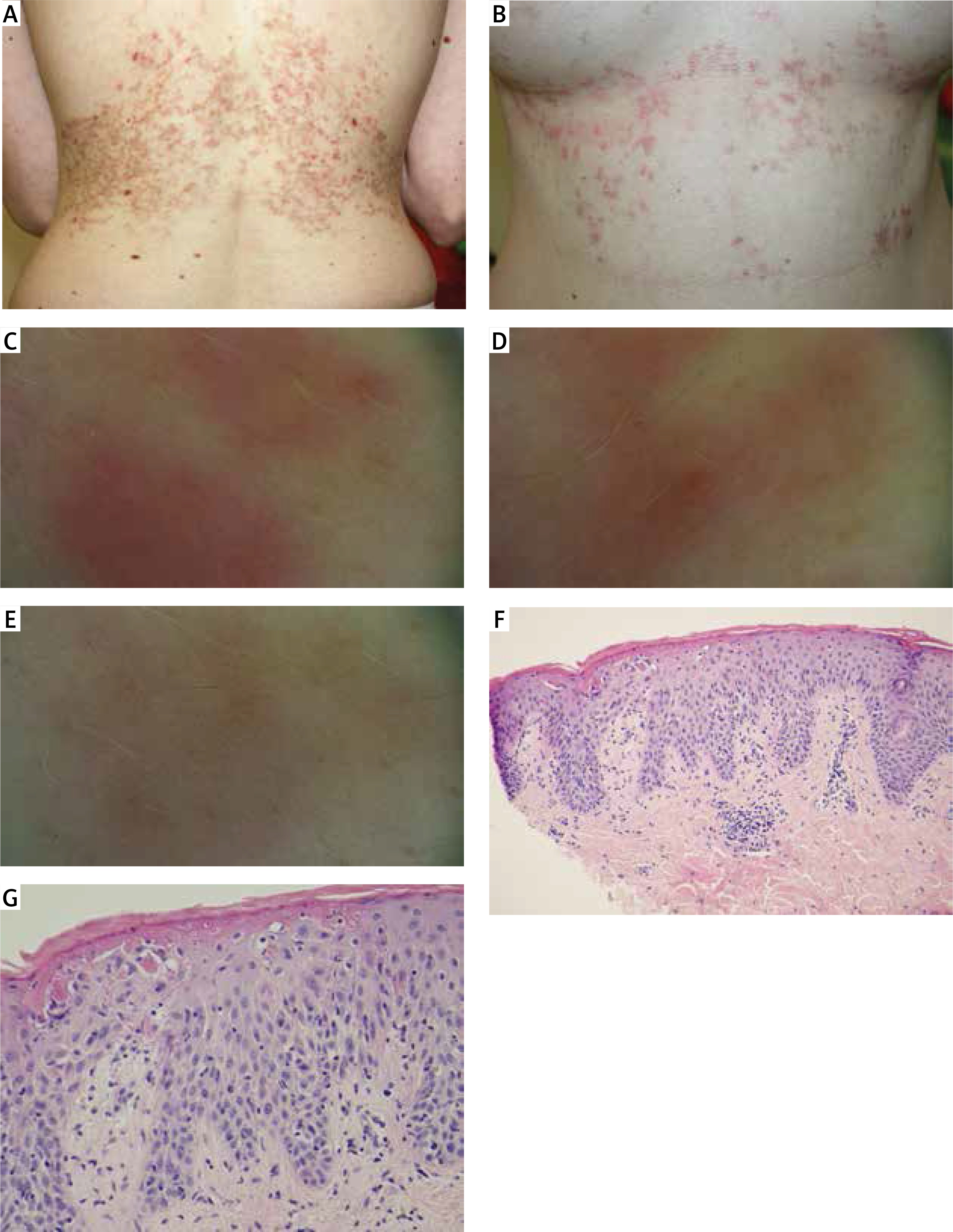
The histopathological test of skin specimens collected from the lesions of abnormal morphology showed:
the first specimen – acute spongiotic dermatitis with eosinophilic infiltration and superficial perivascular inflammation,
the second specimen – acanthosis, exocytosis of lymphocytes, loosening of attachments between keratinocytes, impaired keratosis with dense keratin, parakeratosis and dyskeratosis in the upper part of the epithelium, and superficial serum microvesicles. In the dermis a perivascular infiltrate was observed (composed of lymphocytes, monocytes and rare eosinophils) as well as sparse melanophages (Figures 1 F, G).
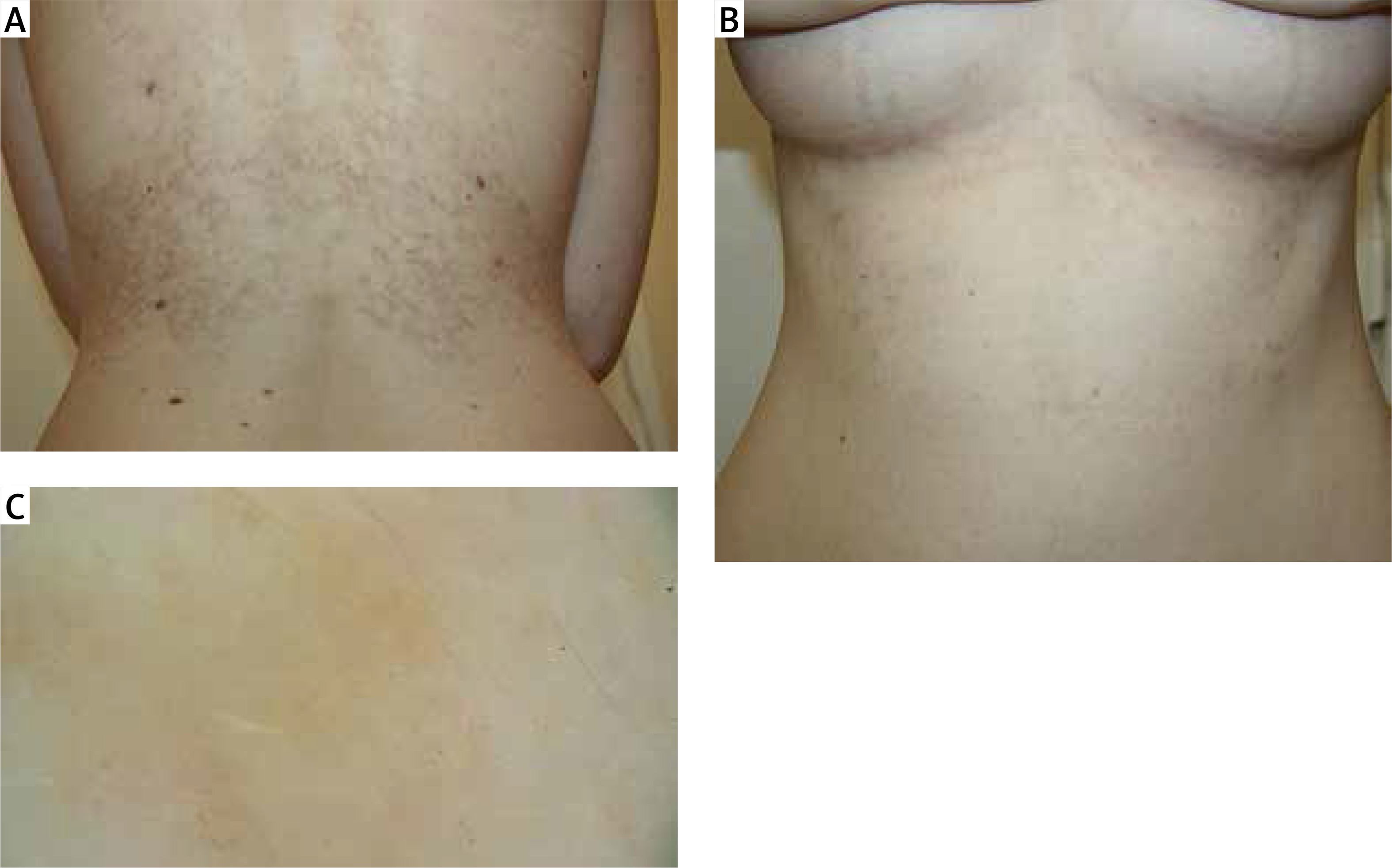
Based on the clinical picture and the histopathological image the patient was diagnosed with PP and the treatment with doxycycline of 200 mg daily was introduced. After 6 weeks of treatment improvement was observed – a complete remission of active foci as well as the pruritus and the burning sensation – the patient described their intensity as 0 out of 10 on the VAS; hyperpigmentation only remained on the skin (Figures 2 A–C).
Figure 2
Photographs of the patient following the treatment with doxycycline: remission of active lesions, persisting fuscous and brown patchy hyperpigmentation: A – back, B – front, C – dermoscopic image: postinflammatory hyperpigmentation in the form of hemosiderin deposits
Although this is the first description of this condition in Poland and in Central and Eastern Europe, it cannot be assumed that this disease does not occur in this region. It appears that PP is rarely found outside Japan as a result of underdiagnosis rather than the less frequent incidence of this condition. The main reason is a diverse histopathological image of lesions of abnormal morphology in individual stages of the disease.







