
Introduction
Sanfilippo syndrome (mucopolysaccharidosis type III (MPS III)), is a rare, inherited metabolic disorder belonging to the lysosomal storage disorder (LSD) group and classified as MPS [1]. The clinical onset of MPS III is uncertain. Patients are born without symptoms and mild to moderate facial dysmorphology and developmental delay may be the first noticeable findings. In the later stages, frequent signs of infection (diarrhea and respiratory tract infections) are added to these symptoms. Since these nonspecific symptoms can be seen in many diseases, it is difficult to suspect Sanfilippo syndrome. Motor function disorders, bone and joint deformities develop within the first few years after birth. Behavioral disorders and communication loss, along with cognitive decline, dominate the picture [1, 2]. Death usually occurs in the second or third decade of life due to neurological disorders or respiratory infections [1, 3, 4].
This article discusses a rare case of an 18-year-old female patient with Sanfilippo syndrome (MPS type III) with concomitant immunoglobulin G (IgG) subgroup deficiency. Permission was obtained from the family for this case presentation.
Case report
An 18-year-old female patient diagnosed with Sanflippo syndrome, epilepsy, and hypothyroidism was admitted to our service. She was also followed by the pediatric metabolism, neurology, and chest disease departments. The patient first came to our outpatient clinic complaining of postnasal drip and recurrent upper respiratory tract infections. The patient’s medical history included frequent hospitalizations due to infections and intravenous antibiotic treatment. In particular, frequent diarrhea, throat infections, recurrent coughing, and frequent inhaler treatment over the past 3 years were noteworthy. The patient had a history of adenoidectomy and previous tests showed growth of Staphylococcus aureus and Streptococcus pneumoniae in sputum culture.
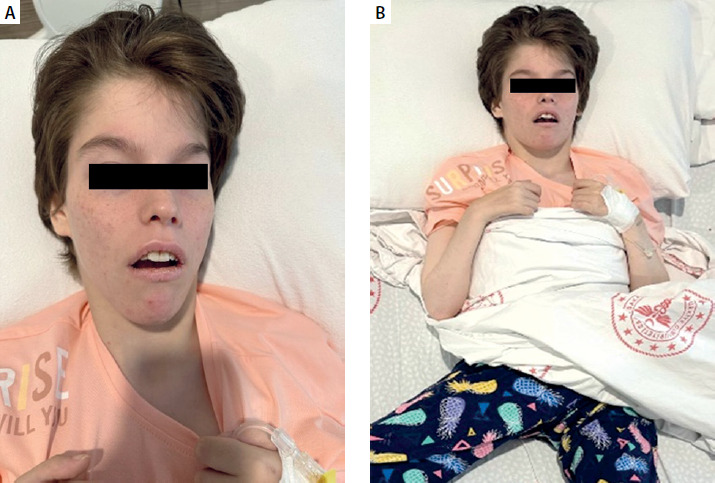
On physical examination, the patient had a neuromotor retarded appearance. She was conscious but not coherent. The patient was quadriplegic with widespread joint contractures in the upper and lower extremities (Figure 1). Bilateral subcrepitant rales were heard by listening in the lungs. There were no other pathologic findings on systemic examination.
Figure 1
Quadriplegic appearance with widespread joint contractures in the upper and lower extremities in our patient
The patient’s complete blood count and routine biochemistry were within normal ranges. In vitro tests showed sIgE levels of < 0.10 for mold, grass, dust, mites, and tree pollen panels, and the inhaler panel. The skin prick test including main foods and inhalant allergens performed at the same time was negative. Serum immunoglobulin test results and flow cytometric evaluation of lymphocyte subsets are shown in Tables 1 and 2. Due to low IgG1, IgG3, and IgG4 levels in the patient’s tests and severe clinical symptoms, intravenous immunoglobulin (IVIG) treatment was initiated with a diagnosis of IgG subclass deficiency. The patient’s frequent upper and lower respiratory tract infection symptoms following IVIG treatment have subsided. The patient was receiving IVIG treatment for 10 months, and during this period, there was a decrease in hospital admissions, infection frequency, and inhaler use. She is also receiving daily physiotherapy, different anti-epileptic medications and symptomatic treatment including inhaled corticosteroids.
Table 1
Serum immunoglobulins of our patient
Table 2
Lymphocyte subsets of our patient
Discussion
Upper and lower respiratory tract infections are common in disease groups in a group of mucopolysaccharidoses. Respiratory problems may also develop due to neurological problems, inadequate mucus drainage, and anatomical airway obstruction [1, 4]. Sanfilippo syndrome (MPS type 3), although defined as a classic neurodegenerative MPS type, has recently attracted attention due to its association with the immune system.
Recent research on MPS suggests that glycosaminoglycans (GAGs) may function as potential endogenous danger-associated molecular patterns (DAMPs) and therefore trigger the innate immune signaling pathway [5]. Congenital defects in immune response have been identified in many types of MPS. Lysosomes are important not only for macromolecule degradation but also for immune functions such as phagocytic activity, antigen presentation, Toll-like receptor (TLR) signaling, and T-cell activation [5, 6]. When lysosomal antigen processing pathways are disrupted, antigen presentation via MHC II is also disrupted. T-cell activation and B cell functions are affected because they are regulated by antigens processed in lysosomes [6, 7]. This condition predisposes MPS patients to infections and inappropriate inflammatory responses, paving the way for immunodeficiency [8]. In a study conducted by Wiesinger et al. on the use of immunomodulatory drugs in patients with MPS, it is stated that the storage of substances that are not broken down in lysosomes triggers an innate immune response and that this response causes clinical deterioration along with inflammation [9].
The literature reports both immune cell dysfunction and B cell response deficiencies in MPS patients [10]. In a study by Lopes et al., a decrease in NK cells and monocyte levels was observed in patients with MPS VI, along with a decrease in T cytotoxic and T helper cells in the memory state [10]. Our patient’s flow cytometry analysis revealed that T cell subsets, B cells and NK cells were generally within normal reference ranges. However, a mild decrease was noted in percentage of TCRab+ T cells (81.36%; normal range: 87–99.3%). This finding may indicate impairment in the T cell receptor repertoire, suggesting a potential underlying immunodeficiency. The reduced TCRab expression could interfere with effective antigen recognition, potentially compromising the T cell-mediated immune response. This may be associated with the recurrent infections observed in our patient.
This situation has led to a delayed immune response and an increased risk of infection. Increased susceptibility to respiratory tract infections in patients diagnosed with MPS VI has been explained in this way [10]. Studies have also shown that NK cell deficiency or dysfunction is associated with increased susceptibility to pulmonary infections, a fatal complication of MPS [4, 10]. The weak antibody response in MPS patients causes an inadequate response to T cell-independent antigens such as pneumococcal polysaccharides in some MPS subtypes [10, 11].
In line with the literature, our patient also had deficiencies in IgG subclasses (IgG, IgG1, IgG3, IgG4) and frequent respiratory tract infections. After IVIG treatment was initiated, there was a significant reduction in frequent infections and hospital admissions.
Conclusions
Frequent respiratory infections in children diagnosed with MPS may be related not only to mechanical causes but also to immune system involvement [12]. In patients with MPS and recurrent respiratory tract infections, lysosomal dysfunction may impair antigen presentation and immune cell function, leading to IgG and subclass deficiencies. This immunodeficiency increases susceptibility to infections, and significant clinical improvements can be achieved with IVIG treatment in appropriate patients. Immune deficiency can develop in lysosomal storage diseases due to genetic mutations or secondary lysosomal dysfunction [13]. Therefore, immune system functions should be monitored periodically in all children diagnosed with LSD, and additional treatments should be planned when necessary. A multidisciplinary approach is essential for improving the quality of life of these patients.










