
Pulmonary arterial hypertension (PAH) is a rare disease. The prognosis of PAH is usually poor and it is the worst in the case of forms associated with connective tissue diseases. In recent years we have observed an improvement in the effects of PAH treatment.
Systemic sclerosis (SS) is an autoimmune disease. The clinical symptoms include generalized hardening of the skin, extensive lesions in visceral organs, including the gastrointestinal tract, heart, lungs and kidneys. The CREST syndrome (calcinosis, Raynaud, oesophageal dysmotility, sclerodactyly and telangiectasia (5–12%)) develops pulmonary arterial hypertension associated with specific diseases (A-PAH) secondary to interstitial pulmonary fibrosis or isolated pulmonary arteriopathy, and pulmonary hypertension (PH) associated with left heart involvement [1–3].
Myxoma is a benign neoplasm of the heart and constitutes approximately 50% of all benign neoplasms of the heart [4]. In approximately 75–80% of cases a myxoma is located in the left atrium (LA), in 20–25% of cases in the right one. The clinical symptoms associated with the myxoma can be divided to systemic ones and resulting from the presence of the tumour in heart chambers. Systemic symptoms include decreased body mass, elevated body temperature, skin lesions, musculo-articular pain, increased C-reactive protein (CRP) and immunoglobulin levels [5–7]. Presence of antinuclear antibodies and the rheumatoid factor can be observed as well. In the medical literature these symptoms are referred to as a myxoma syndrome [8].
Cardiovascular symptoms result from disturbances in the heart function caused by the tumour. Systemic symptoms and abnormalities on laboratory tests observed in myxoma are similar to those observed in inflammatory diseases, including those of autoimmune origin, such as SS. It is presupposed that interleukin 6 (IL-6) is mainly responsible for this fact. Interleukin-6 is a cytokine modifying the inflammatory response by affecting the differentiation of T lymphocytes and transformation of B lymphocytes to plasmocytes, and stimulating the liver to produce acute-phase proteins. Hirano et al. found out that myxoma cells produce IL-6 [9]. An increased concentration of this cytokine is observed in 80% of patients with diagnosed myxoma. Interleukin-6 cytokine concentration is currently a valuable marker in the diagnostic process and post-operative monitoring of the tumour [10].
In PAH, cytokines such as IL-1, IL-6, IL-8 and tumor necrosis factor α (TNF-α) play a significant role in the pathogenesis. It has been shown that in patients with PH endothelial-to-mesenchymal transition (EndMT) takes place. These cells produce significant quantities of pro-inflammatory cytokines and have a similar phenotype to pulmonary fibroblasts of patients with SS [11, 12].
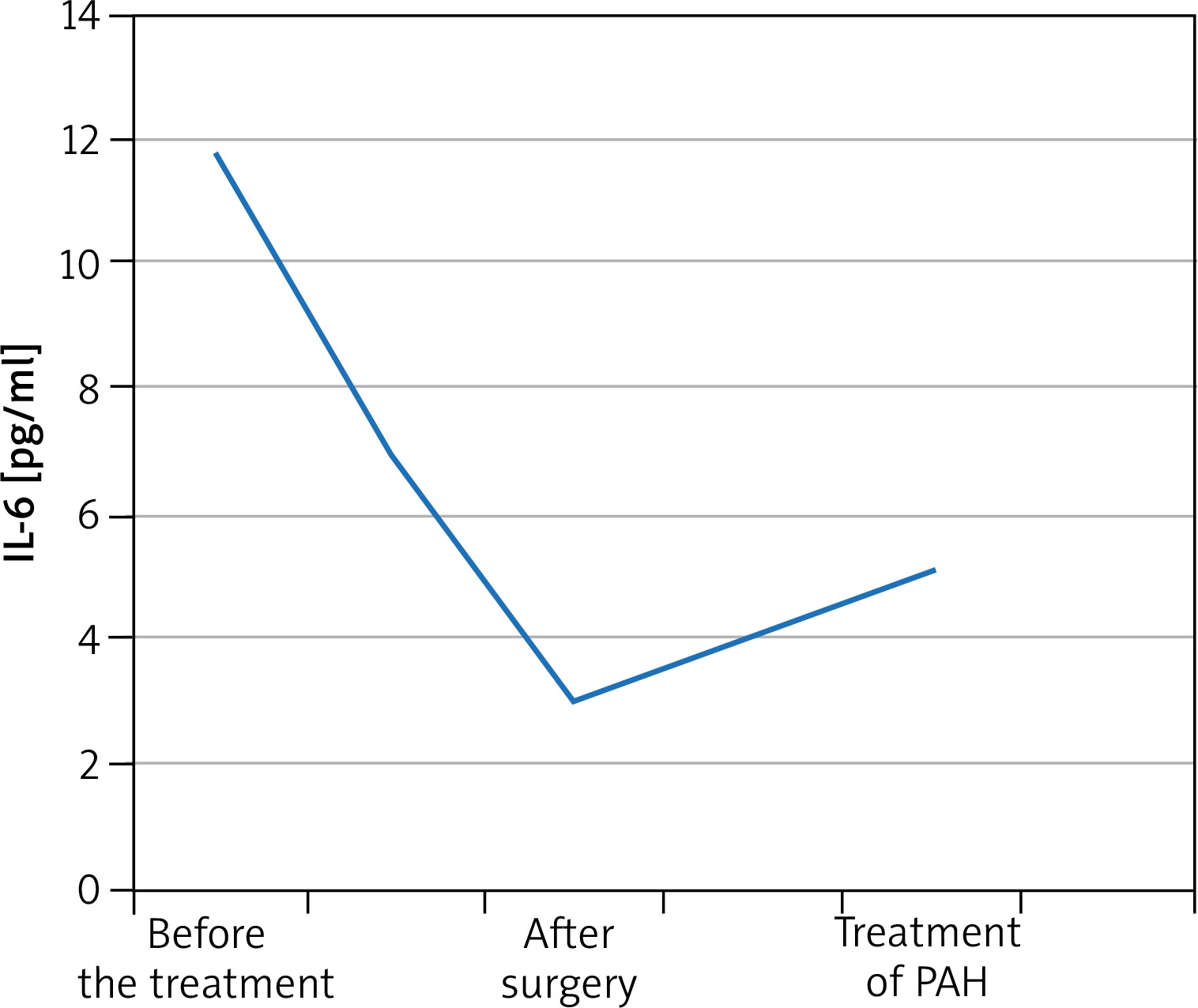
A 55-year-old male patient was admitted to the Department of Rheumatology for diagnostic evaluation due to Raynaud syndrome and a sensation of too tight skin on hands and feet. The symptoms were accompanied by reduced tolerance to physical exertion, pain in the epigastric fossa, heartburn, belching and body mass loss. On physical examination we observed Raynaud sign, scars and acute erosions on the skin of the fingers, brown skin coloration, hard skin in distal areas of hands and feet and a quiet diastolic murmur over the tricuspid valve. Laboratory tests revealed erythrocyte sedimentation rate (ESR) 28 mm/h, reticulocytosis 56‰ (norm: 5‰), microalbuminuria 0.65 mg/l (norm: 0–20 mg/l), and creatine phosphokinase 338 U/l (norm: 38–174 U). Anti-nuclear antibodies (ANA) in titres of 1 : 2560 of a fine speckled pattern were found. There were no anti-centromere antibodies (ACA), anti-topoisomerase antibodies (scl-70), anti-cardiolipin antibodies, anti-endothelial cell antibodies (AECA), cryoglobulins, immune complexes, or anti-neutrophil cytoplasmic antibodies (ANCA). Imaging examinations revealed on oesophageal scintigraphy: disturbances typical of SS, on gastroduodenoscopy: severe esophagitis indicating gastroesophageal reflux disease, on high-resolution computer tomography (HRCT): evidence of interstitial pulmonary inflammation. The cyclic therapy with cyclophosphamide at doses of 1000 mg and with small doses of glucocorticosteroids (methylprednisolone 8 mg/day) was started. Routine echocardiography showed a non-homogeneous tumour of 33 × 51 mm in size in the right atrium (RA), which was pedunculated at the interatrial septum and mobile. The patient had the tumour surgically removed from the atrium and aortocoronary bypasses were implanted. On histopathological examination, a myxoma was diagnosed. The sensation of hard skin decreased, tolerance to physical exertion improved and the dyspeptic symptoms subsided. The patient remained under rheumatological and cardiologic care for another 5 years. After 5 years his general condition deteriorated. He had to be hospitalized due to heart failure (NYHA IV). Echocardiographic examination revealed increased systolic pressure in the right ventricle (110 mm Hg), right ventricle enlargement and reduced pulmonary flow acceleration time (37 ms). Right heart catheterization (RHC) was performed which confirmed PH artery pressure (PAP) 100/40 mm Hg, mean PAP (MPAP) 60 mm Hg. We initiated treatment with treprostinil in a continuous subcutaneous infusion. Treatment of the coronary disease was continued along with rheumatological and pulmonological treatment. The patient’s condition was improved and the clinical condition was stable (NYHA III). However, after one year the patient’s condition deteriorated (NYHA IV class). After another RHC oral treatment with sildenafil was initiated to a satisfactory effect. After another 12 months the patient’s physical efficiency became worse again. Thoracosurgical consultation was conducted to consider lung transplantation (LTx). However, due to numerous comorbidities the patient could not be qualified for LTx. The IL-6 serum level was measured before the surgery and then monitored in the postoperative course. The cytokine concentration before the surgery was 11.7 pg/ml. One month after the surgery, the IL-6 level decreased by almost two times and amounted to 6.9 pg/ml. After 4 months it was 3.0 pg/ml. The IL-6 level was monitored in subsequent months after the cardiosurgical treatment and amounted to 4.3 pg/ml and 3.6 pg/ml. During PAH therapy it amounts to 5.1 pg/ml. The IL-6 level did not grow up to the level from before the operation, but has increased since PAH was diagnosed and started to be treated (Figure 1).
In the described case, during routine diagnostic evaluation for cardiac and pulmonary circulation abnormalities, the patient with SS was found to have a myxoma of the RA. The neoplasm did not cause any cardiovascular symptoms. However, after excision of the myxoma, systemic symptoms started to subside. The sensation of hard skin decreased, tolerance to physical exertion improved and the dyspeptic symptoms subsided. During further observation, progression of the systemic disease was observed and PAH that required a specific therapy. In SS, cytokines IL-2, IL-6 and TNF-α are believed to play a very significant role in the process of vascular damage and initiation of fibrosis [13]. Interleukins are produced by Th2 lymphocytes and non-immune cells, including fibroblasts [13]. Needleman et al. proved that in SS fibroblasts produce approximately 30 times more IL-6 than fibroblasts in healthy people [14]. Also, it was proven that the concentration of interleukin-6 is associated with SS activity and is a prognostic factor for organ lesions, especially in the lungs [15]. The effect of IL-6 is particularly noticeable in patients with SS and diffuse skin involvement observed in our patient [13, 15, 16]. In our case, all three disease entities (SS, myxoma and PAH) were linked by IL-6. An increased IL-6 concentration in a non-specific immune response of the systemic type in the case of a myxoma and auto immune diseases is the source of diagnostic errors [8]. There have been cases described, where cardiac myxomas were initially diagnosed as rheumatological diseases, such as systemic lupus erythematosus, polymyositis, antiphospholipid syndrome or a systemic vasculitis; after tumour removal the symptoms associated with non-specific immune response subsided and the IL-6 level was back to normal [17–19]. It seems interesting whether the symptoms of SS were a manifestation of a coincidence of two completely random diseases or they were connected by a sequence of etiopathogenetic events, and whether IL-6 produced by the myxoma can trigger the onset or enhance the symptoms of SS. On the other hand, we may wonder whether the SS symptoms were not a paraneoplastic syndrome in the course of the myxoma. The situation is even more complicated since in the pathogenesis of PH, IL-6 (along with other cytokines) is produced by modified pulmonary fibroblasts. Thus, it is possible for the PH to be induced not only by the presence of the connective tissue disease itself (SS in this case), but also additionally by presence of a secretorily active myxoma. Surgical treatment of the neoplasm undoubtedly slowed down the progression of the disease as early as in the post-operative course. However, before the discovery of the tumour, the pulmonary tissue was for many years exposed to the effect of cytokines promoting PAH progression. Thus, it is probable that without presence of the myxoma, PH would not have developed at all. What is more, the location of the tumour in the RA could contribute much more to the development of PAH, compared to a location in the left heart. It can be associated with cytokines directly entering the pulmonary circulation and chronic microembolism [20, 21]. While co-morbidity of a myxoma in the LA and PH is relatively commonly described in literature (postcapillary PH), case study reports of co-morbidity or subsequence of a myxoma in the RA and a precapillary PH are very rare. The connection between IL-6 production by a myxoma and the development of PAH has not been analysed either [22]. Moreover, removal of the tumour from the LA most commonly results in subsidence of PH symptoms, while removal of the tumour located in the RA not necessarily causes suppression of PAH progression. This is because a started process of pulmonary circulation remodelling can be irreversible, especially when there are some other factors present that promote PAH progression, such as scleroderma. Presence of structures in the right heart (thrombi, vegetations) usually makes physicians less inclined to apply aggressive surgical management, compared to their presence in the left heart. However, despite its benign nature, cardiac myxoma is characterized by specific biochemical and clinical features described above. Therefore, right-sided location of a myxoma should not be a justification for delaying surgical treatment of the tumour, especially if we take into consideration the risk of PH [21–25].







