
Introduction
Mastocytosis comprises a diverse group of disorders marked by the neoplastic proliferation, expansion, and abnormal accumulation of mast cells (MC) in at least one organ system [1]. There are three main types of cutaneous mastocytosis (CM): maculopapular cutaneous mastocytosis (MPCM), diffuse cutaneous mastocytosis (DCM), and mastocytoma of the skin. DCM is the most serious but the rarest type of CM [2]. This condition is characterized by a generalized erythroderma with a reddish to brown-orange discoloration and extensive bullae. Most cases have blisters that become hemorrhagic with predominance on the trunk and extremities [3]. The clinical symptoms stem from the release of histamine along with other MC mediators such as tryptase, prostaglandins, leukotriene, and cytokines. These mediator-related symptoms include pruritus, blistering, abdominal pain, diarrhea, gastrointestinal hemorrhage, bone pain, and hypotensive episodes. Symptoms range from moderate to severe and in some cases even life-threatening [4, 5].
The diagnosis of CM is based on clinical findings and a skin biopsy [1, 3]. Management includes the avoidance of triggering factors and the use of adrenaline as rescue therapy in cases of anaphylactic shock. Therapies against mast cell mediators such as antihistamines, omalizumab, and mast cell-targeted treatment, which is primarily based on cytoreductive therapy, wherein tyrosine kinase inhibitors (TKI) are employed. Eligibility for imatinib therapy is restricted to patients lacking the KIT D816V mutation, as the presence of this variant confers resistance to tyrosine kinase inhibition [6–8].
OBJECTIVE
We present an unusual and challenging case of a newborn which was excluded from treatment with KIT-TKI due to a mutation in the KIT gene at codon 816. He was treated with vinblastine in 2015 as a part of cytoreductive therapy when new therapeutic options were not yet available.
CASE REPORT
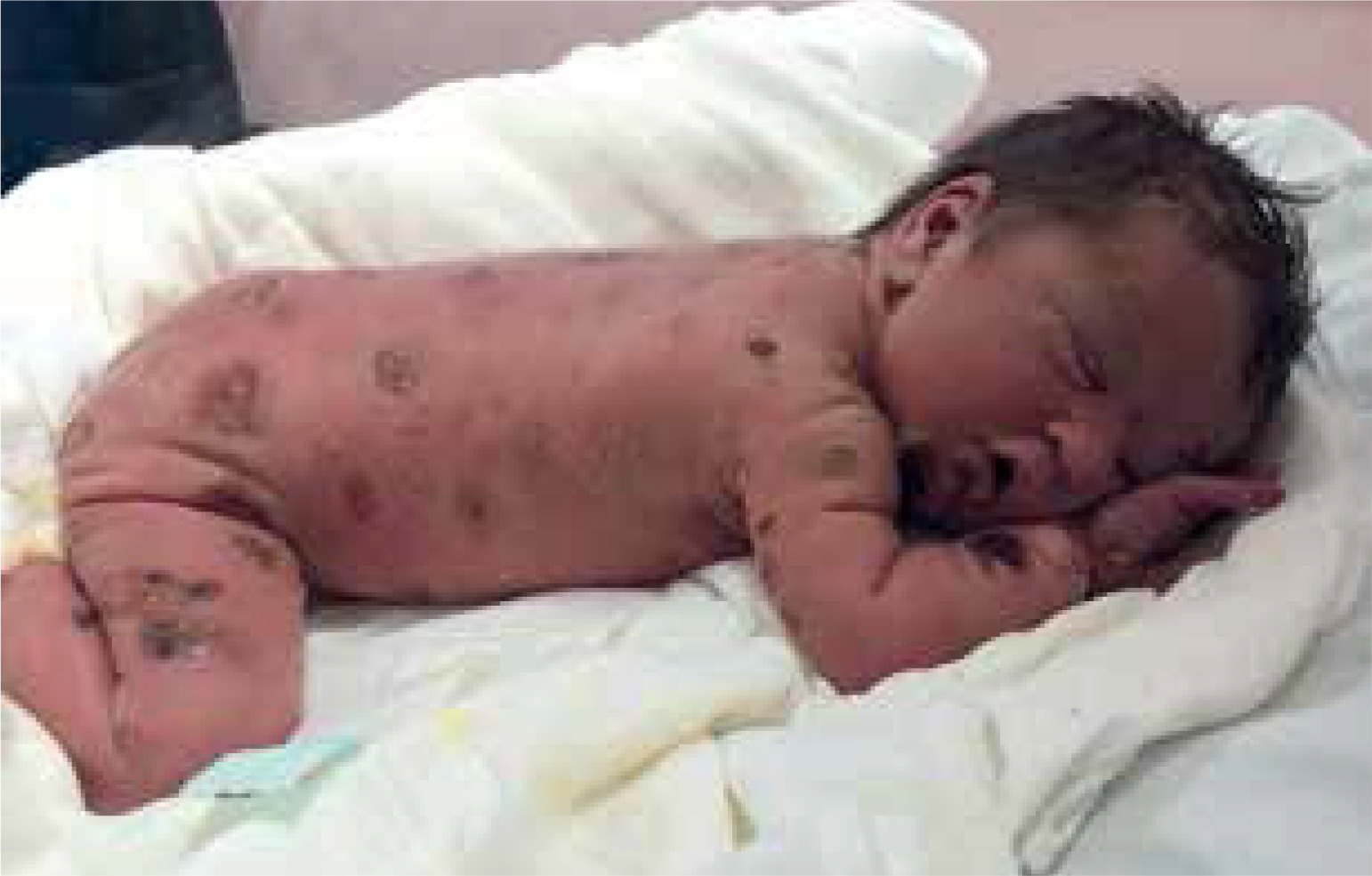
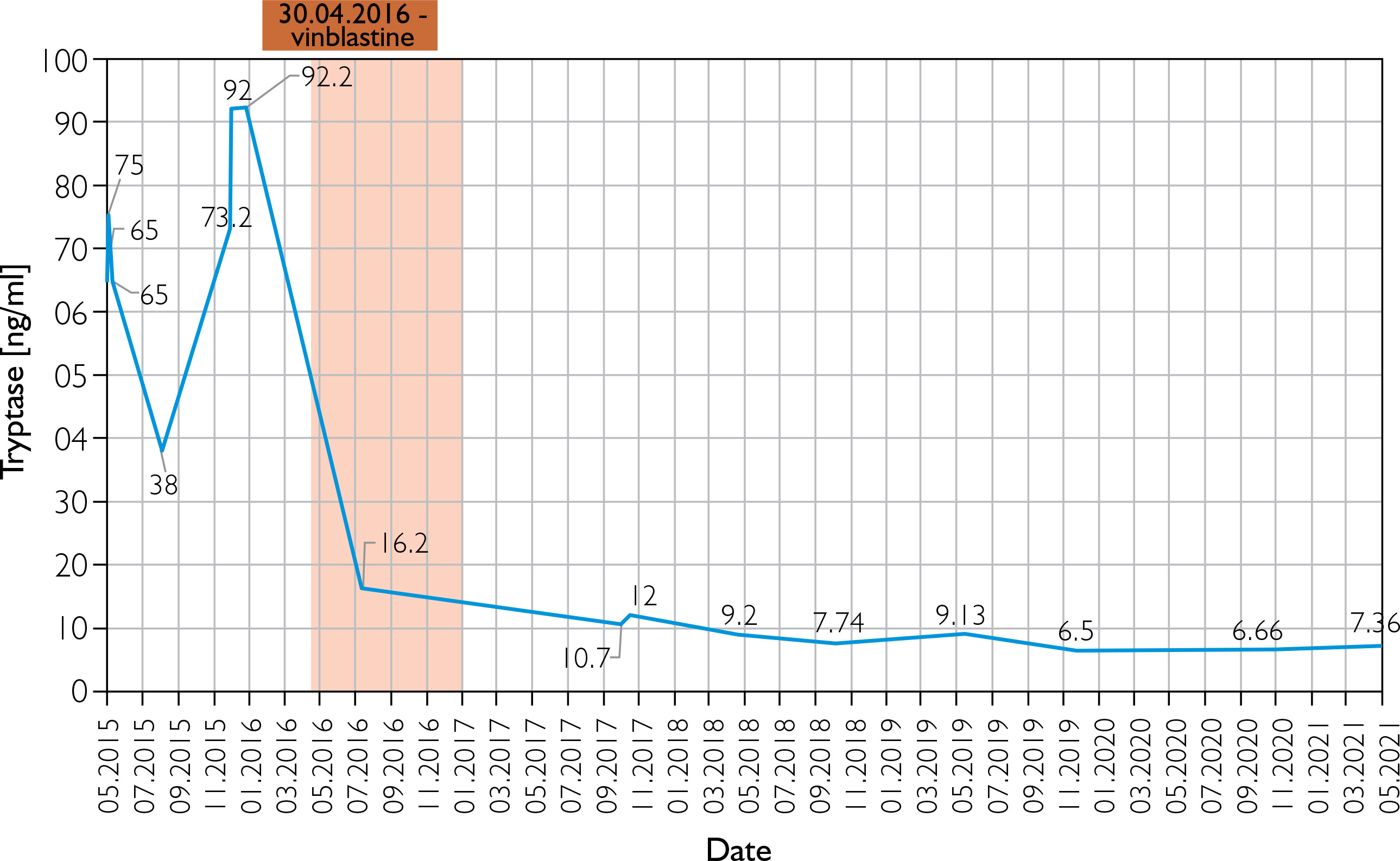
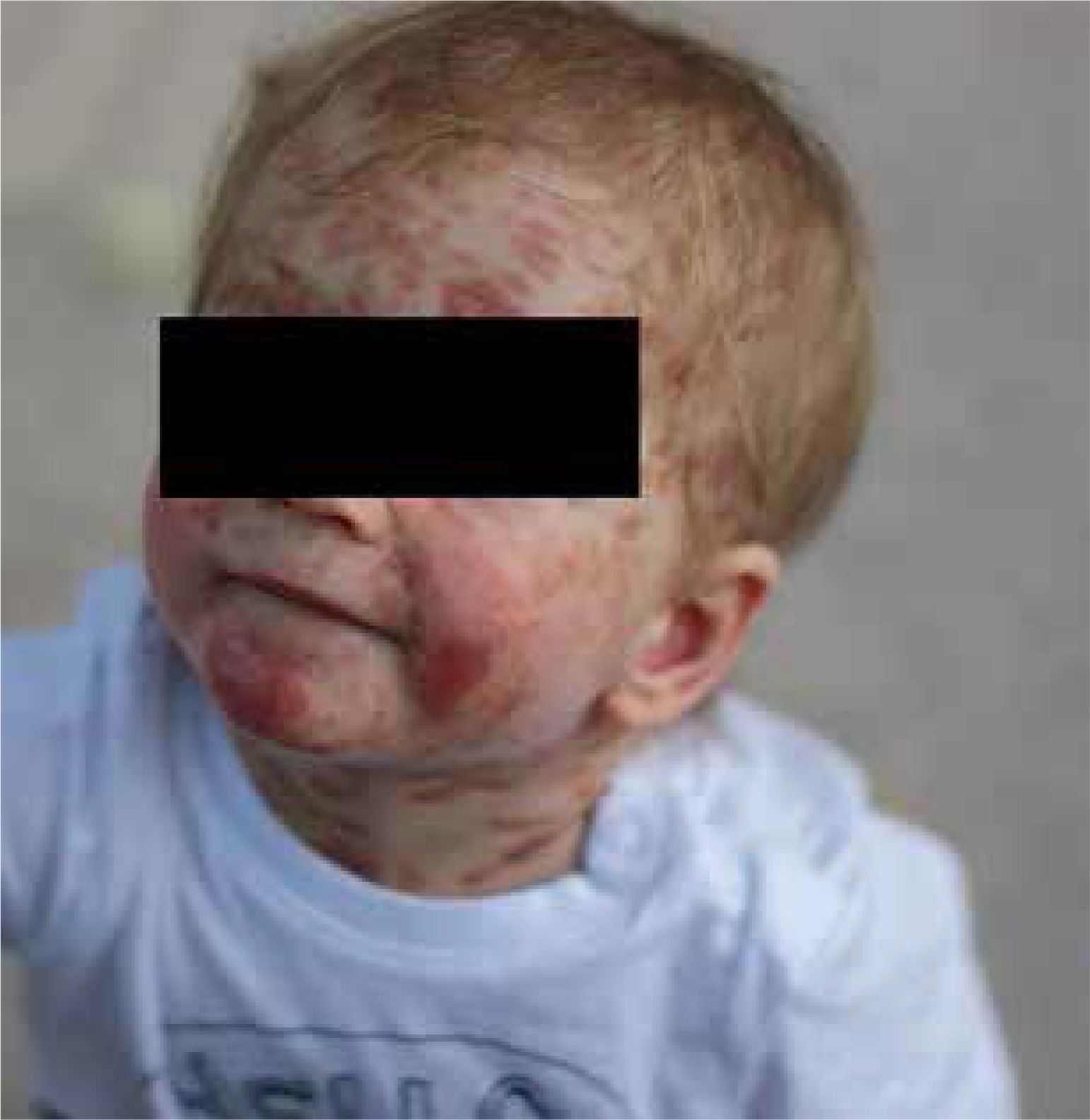
A male newborn was born at 39 weeks gestation, weighing 3120 grams. The patient was delivered by a 29-year-old mother via vaginal delivery. Apgar score was 10. The pregnancy was uncomplicated, with no clinical or laboratory indications of toxoplasmosis or other TORCH (toxoplasmosis, “other” infections such as syphilis and hepatitis B, rubella, cytomegalovirus, and herpes simplex) infections. The neonate presented at birth with multiple hemorrhagic and serous-filled vesicles on the trunk, head, extremities, and scalp (Figure 1). Taking these symptoms into consideration, inherited epidermolysis bullosa (EB) was suspected, and thus empiric prophylactic antimicrobial therapy was introduced. On admission, the boy was in a general good condition, as he did not present with any signs of cardiorespiratory insufficiency. On physical examination, the skin was dry with diffused maculovesicular lesions filled with blood and serous fluid. Cyanotic-pink nodules arranged in garlands were observed on the back. The oral cavity and nails were unremarkable. Results of laboratory and microbiological testing excluded generalized infection and TORCH infections. Blood count with differential was unremarkable, thus excluding hematologic neoplasm. Initially, the patient’s symptoms were manageable, following the protocol used in epidermolysis bullosa acquisita (EBA) symptomatic treatment. The patient showed recurrent episodes of flushing with transient tachycardia of > 200 beats per minute, oxygen desaturation decreased to 80%, and hypotension. Similar episodes were observed during rapid temperature changes, blood tests, and other procedures. After a skin biopsy, the boy had a seizure along with the above-mentioned episodes. Skin lesions evolved with time, in previous locations of the vesicles, nodules were observed, furthermore, generalized thickening of the skin and persistent hyperpigmentation were observed. Due to unclear clinical findings, the neonate was examined by a dermatologist. A histopathological examination of the skin was performed in the first month of life. The histopathological examination with immunohistopathological typing revealed dermal infiltration of mast cells with anti-tyrosine protein kinase Kit (CD117+) and low mitotic count. Based on clinical presentation and skin changes, the following diagnoses were taken into consideration: epidermolysis bullosa, incontinentia pigmenti, and ichthyosis epidermolytica. The definitive diagnosis of CM was made based on immunohistochemical typing. Given the patient’s complex presentation, he was evaluated by multiple dermatologists and referred to a national center for mastocytosis treatment, where the diagnosis of DCM was confirmed. Under these circumstances, a treatment including prednisone (1 mg/kg/day p.o.) and antihistamines was introduced. The treatment was useful in decreasing flushing and tachycardia, however, it did not alleviate skin symptoms. Because of no improvement, a skin biopsy was sent to the Institute of Hematology and Transfusion Medicine in Warsaw which identified a c-KIT mutation in codon 816 (D816V mutation). The mutation excluded our patient from treatment with imatinib. Recurrent hepatosplenomegaly raised suspicion of systemic mastocytosis, therefore a bone marrow sample was examined, however, there was no trace of KIT D816V mutation in bone marrow cells. After 3 weeks of steroid therapy with minimal improvement, an alternative treatment plan was devised. Following consultation with oncologists, the patient received 20 cycles of intravenous vinblastine (4.5 mg/sqm of the body surface area) at 7-day intervals. The therapy was complicated by isolated neutropenia, with total neutrophil count of 0.6 thousand/μl and segmented granulocytes comprising only 17% of the differential. Neutropenia led to a short-term pause in treatment and resolved spontaneously. Mild nausea and vomiting were managed symptomatically. Treatment was completed at the age of 1.5 year, resulting in a significant reduction in serum tryptase levels (as shown in Figure 2) and in the improvement of clinical symptoms. Although skin lesions diminished, they did not completely resolve (Figure 3). The patient had follow-up visits until the age of 6. The psychophysical development during this period was uncomplicated.
DISCUSSION
CM can be classified into three distinct categories [9–14]. The DCM is the least common form [2, 15]. The onset of DCM is typically reported during the first few months of life or at birth [3, 16]. Most patients with DCM exhibit blister formation, presenting as both small vesicular lesions and large hemorrhagic bullae and leaving residual hyperpigmentation which may be the initial manifestation of the disease [2, 3, 15–17]. In infants, elevated serum tryptase levels result from a large infiltration of mast cells over the entire skin and from an unfavorable body surface area to body weight ratio. The underlying issues with the production of mast cell mediators and the accompanying signs and symptoms make it necessary for children with DCM to be admitted to a hospital [15, 17, 18].
In our case, the patient presented at delivery with skin lesions characterized by large hemorrhagic blisters and vesicles, nodules, diffusely indurated skin known as “peau d’orange” and hyperpigmentation. Additionally, the patient demonstrated a markedly increased serum tryptase concentration, consistent with enhanced mast cell degranulation and the associated symptomatology. As a result, the patient was admitted to the hospital and the tryptase level was closely monitored. The general diagnosis is based on the typical morphology of skin lesions, a positive Darier’s sign, and the histology of lesional skin, because mastocytosis mostly presents as a cutaneous disease in children [10, 14]. Our patient met all the requirements for being diagnosed with DCM. The parents of the diagnosed patient were advised to prevent activities potentially inducing mast cells degranulation. Examples of triggers may include mechanical or other types of stimulation, infections, allergens, medications, and exposure to temperature differences. Parents were informed that, in the event of anaphylactic shock, adrenaline should be used. Mediator-directed pharmacotherapy was likewise initiated in the child. Antihistamines, along with oral corticosteroids and pain relievers, are commonly administered to relieve the symptoms of mastocytosis. Currently, there is no specific drug for mastocytosis. Cutaneous mastocytosis often spontaneously subsides.
Children with mastocytosis do not often require cytoreductive therapy, like KIT-targeted tyrosine kinase inhibitors, due to a good chance of improvement without treatment. Nevertheless, some children diagnosed with CM and lacking the KIT mutation at codon 816 have reported a reduction in their skin lesions, when treated with imatinib [6, 9, 19]. The qualification process for mast cell-targeted treatment was initiated in our patient, driven by the considerable severity of skin lesions and associated symptoms. This involves detecting the KIT D816V mutation in peripheral blood through a highly sensitive allele-specific quantitative polymerase chain reaction (AS-qPCR). The examination revealed a mutation in the KIT gene at codon D816V, thereby disqualifying our patient from receiving treatment with imatinib. In this situation, the multidisciplinary team consisting of dermatologists, oncologists, and neonatologists chose to use vinblastine for cytoreductive therapy, identifying it as the best alternative method of treatment. In infantile DCM, the KIT mutation that precludes the use of imatinib is encountered only infrequently.
Vinblastine stands as a prominent alkaloid in cancer treatment, exerting its mechanism of action by binding to tubulin and disrupting the formation of microtubules, working as a mitosis suppressor. This drug finds application in treating various cancers, including those affecting the breast, lungs, head, and neck, as well as Hodgkin’s lymphoma [20]. An unconventional use of chemotherapy has proven effective in alleviating symptoms for the patient, resulting in reduced skin lesions, diminished flushing, and tachycardia. By the age of 8 years, the boy demonstrated progressive improvement in cutaneous symptoms, which were primarily characterized by residual discoloration and hyperpigmentation.
The introduction of kinase inhibitors active against mast cells harboring the D816V and other KIT mutations has substantially advanced the therapeutic management of systemic mastocytosis (SM) [21]. SM is driven by a mutation in KIT (D816V) [22]. In contrast, imatinib is approved by the Food and Drug Administration (FDA) for SM in patients with KIT mutations outside of exon 17, but it is not effective for D816V-associated disease. The drug has been used to treat children with extensive CM accompanied by severe systemic symptoms and blistering, with underlying KIT mutations in exon 8 [9, 11].
In our patient’s case, due to a mutation in the KIT gene at codon 816, imatinib was not a viable treatment option. New therapeutic alternatives with kinase inhibitor properties, such as midostaurin, avapritinib, and nintedanib, have shown promise in SM but were unavailable in 2015 when our patient was treated. The FDA approved midostaurin for the treatment of adult patients with newly diagnosed acute myeloid leukemia (AML) on April 28, 2017. Avapritinib was approved by the FDA in January 2020. In March 2020, nintedanib was approved for use in the treatment of chronic fibrosing interstitial lung diseases. These drugs are currently being investigated for the treatment of patients with mastocytosis and a mutation in the KIT gene at codon 816 [21]. Due to the lack of other therapeutic options at that time in 2015, our patient was treated with vinblastine, and this treatment brought effective results for the symptoms relief. The development of new drugs for the treatment of mastocytosis is a great hope for patients with various mutations in the KIT gene [22]. Five case studies have described the wild variant of KIT in neonatal DCM – three case studies of D816V (p.Asp816Val), dup A502Y503, and Ala502_Tyr503dup in one case study [23–27]. In specific cases, some patients do not undergo genetic testing for mutations in the KIT gene. However, it is advisable to consider additional genetic diagnostics. This becomes especially pertinent in advancing our understanding of the molecular mechanisms associated with DCM and also predicting the future phenotype of the disease, particularly in its more severe manifestations.
CONCLUSIONS
Diagnosis and treatment of DCM in newborns pose a challenge. A holistic approach to the patient and cooperation between a pediatrician, a dermatologist, and an oncologist in the treatment of DCM are crucial to achieving therapeutic success. The presented case provides additional insights into the variability of diagnosis and management of congenital CM. Treatment with vinblastine for neonatal DCM has not been reported before. Further research into targeted therapies for KIT D816V mutations is required to improve treatment outcomes.










