
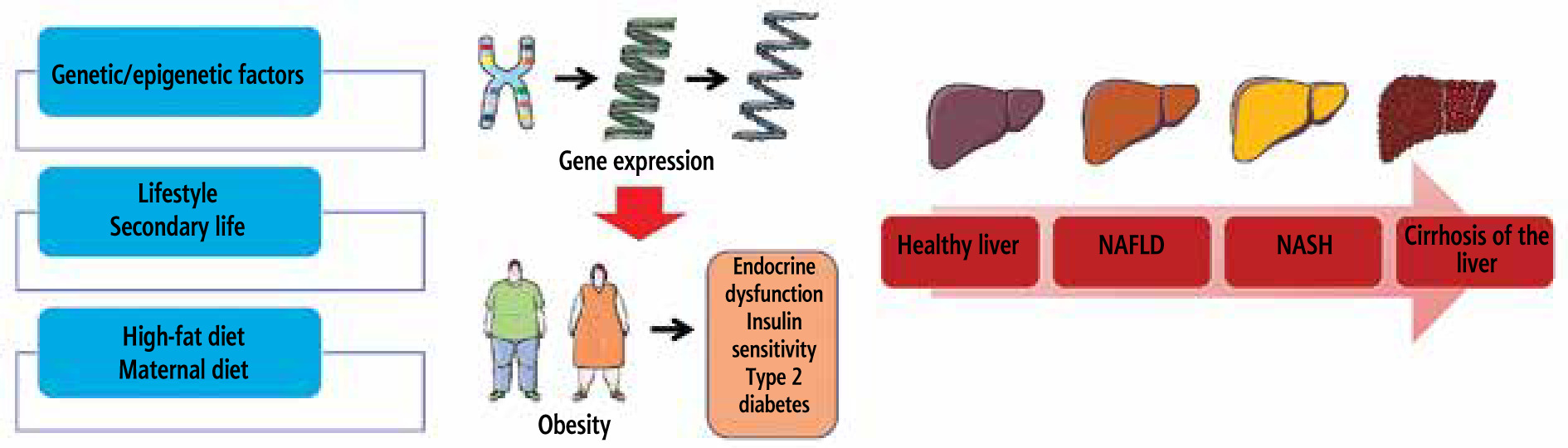
Introduction
A significant public health issue across the world is nonalcoholic fatty liver disease (NAFLD). Several variables contribute to NAFLD, including lifestyle choices, environmental influences, and genetic predisposition. Insulin resistance appears to run in families and is inherited from mothers, suggesting a potential role for genetics in NAFLD. However, environmental variables are likely to influence NAFLD formation significantly, and it seems that a genetic component is also involved [1]. The genotype and quantitative characteristics of each ethnic group by coding the genotype was assessed using an additive inheritance model and evaluating its variance explanation. In obese children and adolescents, there is a correlation between the cytokeratin 18 (CK-18) score and the NAFLD activity score [2]. There is a broader range of disorders that can affect the liver that can be classified as NAFLD. One example of these diseases is nonalcoholic steatohepatitis (NASH), which is a condition characterized by steatosis, cirrhosis, abnormal fibrosis, and steatosis. In addition, hepatocellular cancer belongs to this spectrum [1, 3]. In mitochondria, long-chain fatty acid oxidation is controlled by a protein called carnitine palmitoyltransferase 1 (CPT1). Malonyl-CoA, a metabolite made from glucose involved in making fatty acids (FA) from scratch, stops this enzyme from working. Some researchers showed that preventing steatosis and obesity in mice given a high-fat diet can be simultaneously achieved by chronically elevating CPT1A expression level and a mutant isoform which is insensitive to malonyl-CoA. However, the research did not succeed in correcting the abnormalities caused by the high-fat diet (HFD) in a well-established obesity model [3-5]. NAFLD is not only a “Western illness”; it affects around 15% of the Chinese population. The underlying cause of NAFLD is complex, including interactions between a person’s genes and the environment in which they live. The researchers examined how the polymorphisms of apolipoprotein C3 (APOC3) affected insulin resistance, overweight, fasting triglyceride levels, and total antioxidant status in a Han community in China to better understand their role in risk. The metabolic syndrome, pre-diabetes, and type 2 diabetes are more likely to be developed in about 37% of obese adolescents and children who are overweight. The genetic variation and single nucleotide polymorphism (SNP) associated with alcohol-related cirrhosis and NAFLD in children and adolescents have been linked to these diseases. The membrane-bound O-acyltransferase domain-containing protein 7 (MBOAT7) gene contains the rs641738 gene in overweight children with elevated levels of NAFLD and alanine aminotransferase. The liver phenotype is more significantly associated with the rs626283 SNP, which is cosegregated with rs641738 [6]. The risk of developing a fatty liver is genetically determined at a rate of 40%, with the body mass index (BMI) following a similar pattern. Multiple studies have shown that some SNPs have significant relationships with BMI; however, as yet, no widely shared SNPs with strong impacts on BMI have been discovered across all populations. An acylglycerol lipase with patatin-like phospholipase domain 3 (PNPLA3) transports triacylglycerol and fatty acids across lipid membranes by hydrolysis. Neither its function nor its physiological substrate in liver fat metabolism is well understood. Serum alanine aminotransferase (ALT), liver fat content, and aspartate aminotransferase levels are surrogate markers for NAFLD. In contrast, modifications in PNPLA3 do not have any effect on glucose tolerance, insulin sensitivity, C-reactive protein levels, aminotransferases, or lipid levels. Currently, it is not known whether or not the PNPLA3 rs738409 [G] allele has a direct effect on liver damage or if it is able to optimize the estimation of liver fat and NAFLD by less than 1% [1]. Peroxisome proliferator-activated receptors (PPARs) play a crucial role in metabolic processes involving energy and lipids and are found in brown adipose tissue, liver, heart, and muscles [7]. As PPAR activation suppresses inflammatory responses, it can decrease NASH development from NAFLD, and studies have shown that the PPAR ligand can lower blood lipid levels, increase insulin sensitivity, and decrease liver steatosis. The expression of human PPAR target genes has been examined in many papers; however, due to the difficulties in obtaining biological samples from healthy patients, these investigations have been carried out in small sizes that are not typical [8]. Correctional T1 from magnetic resonance imaging can detect iron and quantify extracellular fluid in the liver, providing a noninvasive method to gauge the severity of steatohepatitis and fibrosis. Phenotyping difficulties have hampered genetic analyses of liver disease susceptibility. The hazards, sample inaccuracy, and variability of a liver biopsy are all factors that make it an invasive operation. An established method in epidemiology, Mendelian randomization, employs genetic research to shed light on causation. Identifying the metabolic characteristics that contribute to steatohepatitis is useful because correcting these factors can significantly reduce the probability of developing liver disease [9].
Material and methods
Scopus provides access to scholarly publications and conference papers in all disciplines, including science, technology, health, the social sciences, and the arts and humanities. Scopus lacks a regulated vocabulary; therefore, the search is limited to either keywords or phrases. The “indexed keywords” that appear in Scopus records are not made up of terms, but rather of topic headings taken directly from the thesaurus of the original dataset from which the reference was extracted. NAFLD was the focus, along with the associated genes.
Results and Discussion
As a result of NAFLD, one can develop liver cancer and cirrhosis. Although there is no cure for NAFLD, early diagnosis and treatment are critical to prevent progression of the disease. There are two key ways to identify genes and mutations associated with NAFLD: genetic testing and mutation screening. Genetic testing can be used to determine whether one is at risk of developing NAFLD, while screening for mutations can help identify which genes are most likely to be involved in the development of the disease.
A healthy diet and regular exercise can reduce the risk of developing NAFLD. Surgery can also be used as a treatment for NAFLD. Medications, such as statins, can also be used to treat NAFLD. Surgery is typically only recommended for patients with severe liver damage. Early diagnosis and treatment of NAFLD can improve the chances of preventing serious health complications.
Overview of NAFLD
There are a number of diseases that are associated with NAFLD, which include steatosis (fatty liver), NASH, and cirrhosis. According to some estimates, anywhere from 25% to 33% of the world’s population has NAFLD, making it the most prevalent chronic liver disease in Western nations. Although insulin resistance, obesity, and other metabolic problems have all been linked to NAFLD, the actual etiology of NAFLD remains unexplained.
Symptoms, causes, and risk factors
Symptoms of NAFLD are uncommon and include, but are not limited to, lethargy, weight loss, stomach discomfort, and jaundice. Obesity, diabetes, high cholesterol, and high triglycerides are risk factors for NAFLD, which has an unclear origin. Hepatocellular carcinoma and cirrhosis are two complications of the more severe type of NAFLD known as NASH. There is no specific treatment for NAFLD; however, lifestyle changes such as weight loss and exercise may help improve symptoms and prevent progression to NASH.
To identify genes and mutations associated with NAFLD, a person must undergo genetic testing. This can be done through a variety of methods, including blood tests, biopsies, and imaging tests. Blood tests can help identify genes that are associated with the disease, while biopsies and imaging tests can help to identify mutations in these genes.
There are several different types of genetic testing that can be used to identify genes and mutations associated with NAFLD. It is most common to sequence the entire genome as a means of genetic testing. This type of testing looks at the entire genome, or all of the DNA, of a person. Whole genome sequencing can be used to identify known and unknown genes and mutations associated with the disease.
Another type of genetic test that can be used to identify genes and mutations associated with nonalcoholic fatty liver disease is called targeted sequencing. This type of testing looks at only specific areas of the genome that are known to be associated with the disease. Targeted sequencing is less likely to identify new genes and mutations associated with the disease than whole genome sequencing, but it can be used to confirm the presence of known genes and mutations.
Screening for mutations
The detection of mutations is an important part of the identification of genes and mutations associated with NAFLD. There are several different ways mutations can be screened for, including blood tests, biopsies, and imaging tests. Blood tests can help identify mutated genes that are associated with the disease, while biopsies and imaging tests can help to confirm the presence of these mutated genes. Once a mutated gene has been identified as associated with NAFLD, it can be screened for in other family members who may be at risk of developing the disease themselves. Screening for mutated genes in family members who do not have the disease can help determine if they are carriers of the mutation, which means that they could pass it on to their children even if they do not develop the disease themselves.
Implications of genes and mutations
The identification of genes and mutations associated with NAFLD can have a number of different implications. First, it can help determine if a person is at risk of developing the disease themselves. It can also help to determine whether they are carriers of the disease, which means they could pass it on to their children even if they do not develop the disease themselves. Finally, the identification of these genes and mutations can also help develop new treatments for the disease.
It was found that NAFLD has been most strongly associated with PNPLA3. Several genes may cause NAFLD, but recent genome-wide association studies (GWAS) have identified PNPLA3 as the first compelling allele related to liver lipid accumulation [1]. And other investigations on liver disease are available in the literature [10-12].
APOC3, or the promoter of protein three that contains the patatin-like phospholipase domain, was not associated with plasma triglyceride (TG) levels of people with nonalcoholic fatty liver disease. However, there could be racial differences in the degree of disequilibrium between the causative variants. In very obese individuals who undergo surgery, the incidence of NAFLD may be more than 90%. The two APOC3 SNPs did not affect BMI, while patients with NAFLD had decreased oxidative stress. In the Han population, neither of the two variations in the APOC3 gene was related to the risk of NAFLD, compared to PNPLA3 [13].
Those with the PNPLA3 148M variant are more likely to develop fatty livers, elevated transaminase levels, and alcohol-induced cirrhosis. As demonstrated in this study, the PNPLA3 148M allele can also increase the likelihood of early liver damage. A sample of overweight and obese children and adolescents was evaluated for liver fat levels using ultrasound imaging. Although it is not the most reliable imaging modality, diagnosis can be made with high reliability with this technique. Liver fibrosis, cirrhosis, and failure are all associated with the PNPLA3 148M allele, suggesting that it could be a useful tool to identify those at risk so that they can receive preventive care and more intensive treatment for other risk factors [14].
An individual with atherogenic dyslipidemia and NAFLD was examined for steatosis of the liver due to rs1051338 SNP in lysosomal acid lipase (LIPA). Rare allele carriers had lower levels of high-density lipoprotein cholesterol (HDL-C), as well as high levels of total cholesterol (TG). In addition to increased levels of ALT, aspartate aminotransferase (AST) and cholesteryl ester transfer protein (CTP), carriers of the uncommon polymorphism allele c.46AC/CC had a higher risk of developing severe liver steatosis (S3). Compared to conventional autosomal recessive forms of lysosomal acid lipase deficiency (LAL-D), attenuated versions with milder clinical symptoms may be caused by the rs1051338 SNP, which was reported to have a loss of function impact on LAL activity. Hepatic steatosis and LAL-D-like dyslipidemia were considerably worse in persons with NAFLD who also had the rs1051338 polymorphism in the LIPA gene [15].
A Greek pediatric population with the rs738409 polymorphism in PNPLA3 was studied to verify whether it is associated with NAFLD and to explore its association with atherosclerosis risk factors. When the authors looked at blood pressure, insulin resistance, and lipid profile, among the other hallmarks of metabolic syndrome, they found no evidence of a connection between the PNPLA3 gene and any of these factors. On the other hand, the presence of both NAFLD and NASH was linked to elevated levels of TG and decreased HDL-C values. Despite the polymorphism rs738409 PNPLA3, carotid intima-media thickness was not associated with subclinical atherosclerosis. However, elevated ALT levels were associated with the rs738409 polymorphism in patients with metabolic syndrome [16].
Regardless of BMI or diabetes status, the adiponutrin genotype rs738409 affects the probability of developing NASH and the severity of existing fibrosis. Extrahepatic comorbidities, including carotid atherosclerosis and chronic renal disease, may be explained by the impact on lipid metabolism. TM6SF2, which encodes a protein that controls liver TG production, has been identified as a disease modifier that may help stratify risk for liver-related morbidity and reduces the output of molecules with a lipid component. According to GWAS, fatty liver disease in Europe is linked to a number of SNPs outside of the gene that produces the glucokinase regulatory protein (GCKR). By controlling glucose uptake and disposal and providing substrates for de novo lipogenesis, the GCKR protein, which is the product of the GCKR gene, is responsible for disrupting the normal balance of glucose and lipids in the body. A recent study using biopsies of more than 600 people suspected of having NASH found that the AA genotype associated with absence of C-mer proto-oncogene tyrosine kinase (MERTK) protects against the development of fibrosis [17].
The UGT1A1*28 genotypes were mixed in the investigation using a dominant model. Although the data did not demonstrate an association between the UGT1A1*28 genotypes and NAFLD in children, more research is needed to determine whether the variant UGT1A1*28 genotype plays a protective function in NAFLD. Liver ultrasound was used in this investigation to identify children with NAFLD in Taiwan. Obese youngsters in Taiwan were shown to have a lower risk of NAFLD if they had the UGT1A1*6 variant genotype [18].
Five genes, FASN, DGAT1, LPL, IRS2, and YWHAZ, were found to be correlated with a poor prognosis in individuals with hepatocellular carcinoma. Regulation of lipid metabolism and the development of liver cancer have been linked to these genes. Reprogramming of lipid metabolism in hepatocellular carcinoma has recently been an area of study. In the light of these observations, it may not be that FASN is responsible for the etiology of HCC, but rather that it is responsible for the prognosis of HCC. Its genetic deletion suppresses Akt-driven HCC growth by inhibiting Rictor/mTORC2 signaling. These findings corroborate a recent discovery that hepatocellular cancer is associated with a change in lipid metabolism. Modulation of SCD and FADS2 enzyme activity in liver cancer cells may be influenced by factors in the tumor environment, such as fibrosis, hypoxia, and dysregulated metabolism [19].
PLIN proteins are essential for proper adipose cell metabolism and have been linked to alcoholic liver disease. According to the findings of their study, the frequency of the PLIN 6 major allele was only slightly more significant than that of healthy controls. There is no correlation between PLIN 4’s minor allele and anthropometric measurements, plasma leptin, glucose, or insulin concentrations; however, variations in PLIN 1 and PLIN 4 genes have been linked to low BMI, poor atherosclerotic and postprandial responses, and metabolic syndrome. Compared to the frequency of the group without NAFLD, PLIN 6 minor alleles were shown to be considerably lower in the NAFLD population. It was in contrast to the population without NAFLD. Given these results, it is plausible that having a low frequency of minor alleles of PLIN 6 contributes to an elevated risk of developing NAFLD [20].
Many genes that control the synthesis or activity of tumor necrosis factor α (TNF-α) have been associated with alcoholic steatohepatitis and NASH. Among them are the endotoxin receptor CD14 and the anti-inflammatory cytokine interleukin 10 (IL-10). Polymorphisms have been detected in the promoter region of the TNF-α gene. Profibrotic growth factor and connective tissue growth factor can be produced by both insulin and glucose in liver stellate cells. A gene variant encoding transforming growth factor β1 (TGF-β1), matrix metalloproteinase 3, or peroxisome proliferator-activated receptor γ (PPAR-γ) may be associated with liver fibrosis, resulting from a variation in genes encoding those proteins. Good study design may help researchers avoid some pitfalls that can arise when using current technology to identify genetic factors associated with the risk of NASH. To conduct GWAS for NASH, a comprehensive SNP-based haplotype map of the human genome must be made available [21].
In people and animal models with NAFLD, the authors discovered that higher liver fat levels are linked to increased insulin resistance. Scientists have found that transgenic mice overexpressing human apolipoprotein C3 are more likely to develop liver steatosis and liver insulin resistance. This results from greater sensitivity to developing these conditions. There is an increase in the amount of diacylglycerol found in the liver, which is associated with these alterations [22].
NAFLD patients have an increased risk of insulin resistance, TNF-α, portal inflammation, and liver fibrosis, all of which have been linked to a polymorphism in the TNF-α gene at position –308. Lower levels of PPAR-γ have been associated with increased adipocyte differentiation, which in turn causes insulin resistance. The progression of fatty liver disease into NASH is influenced by TNF-α. However, human TNF-α studies have produced conflicting findings, which may be due to different inclusion criteria and heterogeneity among research. In addition, some effects of a person’s genes could be unique to a particular population. Patients with fatty liver had a considerably higher frequency of the 238 TNF-α polymorphism compared to healthy controls.
In comparison, patients with NASH had a significantly higher frequency of 1031 and 863A polymorphisms compared to fatty liver. Patients with mutant genotypes are more likely to have moderate to severe portal inflammation and fibrosis than those with wild-type genotypes. The potential biochemical link is insulin resistance [23].
The work of Calvert and colleagues identified possible genes that may be implicated in NAFLD/NASH using bioinformatics and analysis of the gene/protein expression matrix. The authors used a novel protein microarray method called reverse phase protein microarray (RPPA) to investigate the cellular signaling network in NAFLD. Most of this network is regulated by post-translational phosphorylation through the fast enzymatic catalysis of kinases and their respective substrates [24].
Alanine aminotransferase level, total cholesterol level, and family history predicted the prevalence of NAFLD. There is an association between liver steatosis and the rs738409 SNP in the PNPLA3 gene in both adults and children; however, logistic regression analysis was unable to show the independent contribution of homozygosity for the PNPLA3 polymorphism to the etiology of NAFLD. In this investigation, elevated ALT and total cholesterol levels indicated juvenile NAFLD. Due to the possibility that up to 12-29% of children may have regular ALT readings, ALT alone cannot be used to diagnose NAFLD. Although the research only had a small amount of histological data, it was still possible to conclude that NAFLD in children may progress to more severe stages of liver fibrosis. Cases of steatosis and NAFLD can be predicted using ultrasound transient elastography (TE). CAP is a noninvasive measurement that can be used to diagnose and quantify adult steatosis. According to a study, NAFLD communities are associated with high familial prevalence of metabolic diseases, and overweight or obese children are more likely to develop NAFLD if they have a family history of NAFLD and have abnormal laboratory results [25].
Until now, the polymorphism of the NPY gene has not been shown to affect liver histology in participants with NAFLD, but this study is the first to provide such evidence. There was no correlation between BMI, age, and sex and the effect on lobulillar inflammation and steatohepatitis. The authors found a link between rs16147 and liver histology, although the underlying molecular pathways are not evident. It is possible, however, that NPY’s impact on energy expenditure affects how much fat the body stores, particularly in the liver. Additional research is needed to identify whether it is necessary to assess various polymorphisms in the patient with NAFLD at the time of diagnosis and before starting therapy. However, the authors know that the NPY gene polymorphism rs16147 is independently related to a reduced proportion of steatohepatitis and lobulillar inflammation in obese people with biopsy-verified NAFLD [26].
High-protein eaters, especially NASH patients, had better gene expression profiles, but supplementation with n-3 polyunsaturated fatty acids (PUFA) did not improve gene expression or histology. If the dosage is too low, there will be an impact. The limited sample size and cross-sectional nature of this research are two of its potential weaknesses. Although the authors found that n-3 PUFA supplementation did not successfully modulate gene expression or improve liver histology, they did find that several modifiable lifestyle variables, including BMI, were associated with liver gene expression [27].
Here, scientists reported the discovery of a group of genes that are specifically up-regulated in the livers of obese adults due to the presence of thyroid hormones. This group of genes controls most of the processes involved in the metabolism of ribonucleic acids (RNA), protein breakdown, and energy metabolism. The genes that govern RNA metabolism, protein breakdown, and energy metabolism are favorably regulated by T3 in human skeletal muscle. Obese mice on a high-fat diet also have a reduction in these genes. In terms of state-of-the-art research, there have been no previous investigations of how thyroid hormones influence gene expression in the human liver.
Genes involved in obesity and NAFLD
In addition, the researchers outlined a successful approach to gene therapy that may reduce HFD-induced obesity in mice while improving body weight, glucose tolerance, liver steatosis, and hepatic insulin signaling. The benefits of increasing fatty acid oxidation (FAO) in hepatocytes are discussed, and the molecular mechanisms underlying these benefits are elucidated. After comparing lipid profiles in mice and humans, the scientists proposed a circulating biomarker to monitor obesity and liver steatosis and assess the success of therapeutic measures. Their findings suggest treating NAFLD and other obesity-related illnesses by targeting the human version of CPT1A. In the end, the researchers showed for the first time that liver hCPT1AM gene therapy may improve HFD-induced abnormalities in a mouse model of obesity, diabetes, and NAFLD. Furthermore, they found that TAG (C50:1) showed promise as a noninvasive biomarker for monitoring NAFLD in mice and humans [3].
The expression of the CPT1A gene was observed to be positively correlated with the evaluation of insulin resistance homeostasis model (HOMA-IR) in the fatty liver in the obesity group, with a 2.46-fold increase compared to the control group (CN). A vital enzyme in metabolically active tissue, CPT1A catabolizes free fatty acids (FFA) in the mitochondria. Glucose metabolism is controlled by PDK4, which acts as a gatekeeper regulator. There is an inverse relationship between increased expression of PDK4 in peripheral blood mononuclear cells (PBMC) of the obese group (OF), abdominal circumference (AC), and BMI, and there is a positive relationship between increased expression of PDK4 with blood glucose. Researchers have discovered a connection between fatty liver and the genes SLC25A20, ACADVL, and ACAA2. Blood sugar, insulin level, and HOMA-IR are inversely correlated with SLC25A20 and ACADVL, but positively correlated with ACAA2 in the OF group [8].
Several scientists performed a genome-wide association analysis in Han Chinese subjects to assess the risk of NAFLD. The findings indicate that Asians have a greater genetic vulnerability to NAFLD than Europeans and Africans due to the PPARGC1A rs8192678 risk allele. Obesity and insulin resistance are associated with the rs8192678 SNP in PPARGC1A. Even after considering BMI in the multivariate analysis, the risk allele was still able to function as an independent predictor. Liver ultrasound was used to diagnose NAFLD in their institution. To ensure the reliability of their positive relationship results, the researchers used a high threshold of 7. They set the cut-off point for pediatric NAFLD at an ultrasound score of 4, which is relatively limiting. The findings showed that the PPARGC1A rs8192678 risk allele increased the likelihood of juvenile NAFLD in obese children from Taiwan [28].
Existing and previous research has shown that none of the proposed mechanisms for the development of fatty liver apply to individuals with deletions in the Laron syndrome (LS) and growth hormone (GH) genes. Patients with LS and GH gene deficits due to GH gene deletion are characterized by extreme obesity, particularly central visceral adiposity. Fatty liver disease is common among this population; however, it does not affect everyone. All triglyceride measurements were within normal limits, and three of the male LS patients diagnosed with NAFLD also had cholesterol readings within normal limits [29].
Within 24 hours, 48 hours, and 72 hours, cells cultured in 50% fetal bovine serum developed steatosis, which resulted in elevated levels of AST, ALT, TG, alkaline phosphatase (ALP), and high density lipoproteins (HDL). Expression of fat mass and obesity associated (FTO) and fork head transcription factor O1 (FoxO1) messenger ribonucleic acid (mRNA) was more significant in the experimental group than in the control group. Researchers have found that FoxO1 is a critical transcription factor for AMP-activated protein kinase (AMPK) and that activation of downstream metabolic mediators, such as AMPK, may be related to obesity linked to FTO variables. However, it has not been established whether FTO expression is related to FoxO1. Lipidic droplets and fatty tissue are only two of the numerous tissues that express the FTO gene (16q12 a q24). Type 2 diabetes and other endocrine and metabolic disorders have been found to be associated with it. One of the first indicators of type 2 diabetes, especially in obese patients, is low levels of methylation in the FTO gene. Also, more and more evidence points to a connection between FTO and NAFLD [30].
We found that fatty liver, like NAFLD tissue, had elevated percentages of memory T cells and increased Th1/Th17 immune cell phenotypes. Progression of NAFLD, including the onset of insulin resistance and type 2 diabetes, is probably caused by these cells. Both the number of natural killer cells (NK) in the liver and the number of NK cells in peripheral blood were shown to be lower in obese people, and this trend worsened in NAFLD samples. There are two hypothesized mechanisms by which NK cells regulate immunological activity: cytotoxic activity and polarization of resident and recruited macrophages from the circulation. Memory B cell subsets have not been well identified in obesity compared to T cells, NK cells, and macrophages. Studies have shown that antigen-producing plasmablasts and resting memory B cells increase in number and activity in obese people, while the opposite is true for B cells [31].
Several gut hormones have been studied for their roles in obesity. One of these is ghrelin, which has been found to have the opposite effect on the brain, with positive correlations with activation in the prefrontal cortex, amygdala, and insula and negative correlations with activation in the hypothalamus. Although the full scope of intestinal-derived peptides has yet to be determined, recent studies have shown that ghrelin, through the growth hormone secretagogue receptor (GHS-R), can influence both the innate and adaptive immune systems, hence affecting the inflammatory state seen in obesity and NAFLD. In the past 10 years, the intricate relationships between many body systems in obesity and NAFLD have become a focus, along with the need to understand this multifaceted environment to effectively manage health problems [32].
North Indian Asians with NAFLD were studied for PPAR polymorphisms, and it was found that two of the SNPs were strongly linked to the disease. Polymorphisms in the apolipoprotein C3 gene (APO-C3) may be related to NAFLD among Asian Indians, who have a higher proportion of body fat, abdominal obesity, and insulin resistance than white Caucasians. The PPAR gene has been implicated in the development of NAFLD in several studies, despite limited study of the genotype-liver steatosis relationship [33].
However, research on hypothyroid mice reveals that thyroid hormone has different acute and long-term impacts on subsets of target genes. There is only limited overlap between the acute effects of T3 and the gene set that is differently regulated in obesity, suggesting that obesity may be the consequence of deficiencies in thyroid hormone function. In a considerable population investigation, decreasing thyroid hormone levels were positively related to BMI and liver enzymes in euthyroid participants, indicating a relationship between thyroid function and pro-inflammatory responses [34].
It is unclear whether the PNPLA3 rs738409 genotypes alter ALT and AST levels; however, researchers observed a strong link between increased liver fat substance and the PNPLA3 rs738409 genotypes. A multivariate linear regression study of the PNPLA3 gene family showed that specific genotypes of the variation rs738409 were associated with elevated mean blood ALT levels. They found no changes in insulin resistance or obesity between the three genotypes of the PNPLA3 rs738409 polymorphisms [35].
Gene variations and mutations and NAFLD
Some of the most significant odds ratios (OR) to date for impacting NASH/fibrosis and HCC are found in PNPLA3 and three of the four novel NAFLD-associated variations; these variants may soon be integrated into clinical algorithms to predict advanced liver disease. Predicting the prevalence of NAFLD based on preexisting metabolic disorders has limits, as the condition has already been identified. The prevalence of NAFLD can be expected genetically, although this may be understating the power of genetic studies. To predict NAFLD in individuals of Mexican ancestry who are morbidly obese, a genetic risk score was created using variants in or near PNPLA3, TM6SF2, GCKR, LYPLAL1, and PPP1R3B. NASH was not predicted based on the genetic risk score. The genetic variant PNPLA3 I148M, in combination with environmental factors, can lead to the development of chronic liver disease. It may predict NAFLD and provide personalized suggestions to reduce risk. Those who have the I148M allele in PNPLA3 have a better chance of preventing liver disease if they avoid obesity, alcohol, and any other factors that contribute to the development of the disease. However, hyper- or hypofunctional TM6SF2 can increase or decrease susceptibility to dyslipidemia/cardiovascular disease and NAFLD/advanced liver disease/hepatocellular carcinoma, respectively [36].
Fig. 1
How fatty liver disease develops and what variables increase the likelihood of developing NAFLD and NASH
The balance of liver energy was not affected by fat mass or depot distribution in early stage type 1 diabetes. Researchers found higher levels of fetuin in type 1 diabetes mouse models. This was in contrast to lower levels of FGF21, a regulator of mitochondrial oxidative phosphorylation. There is no evidence that the variation in serine encoding of PPARGC1A is associated with higher levels of liver adenosine triphosphate (ATP). However, this variant has been associated with increased risk of obesity and oxidative stress. These results suggest that defensive gene variants of type 2 diabetes may explain why non-obese people with type 1 diabetes who have acceptable liver sensitivity to insulin do not experience an increase in liver ATP dosages. Gene abnormalities in oxidative metabolism are responsible for low levels of ATP in the liver in people with type 1 diabetes [37].
The whole meta-analysis aimed to investigate the connection between the PNPLA3 I148M polymorphism and NAFLD in children and adolescents and quantify the influence of this variation on metabolic comorbidities. Particular attention must be paid to exercising extreme care concerning the following issues: Most of the research was conducted in affluent nations, there was a lack of variety in terms of ethnicity, and the results might have been skewed by factors such as the author’s bias or experimental inaccuracies. Patients may benefit the most from early lifestyle modifications, which may be significantly facilitated by early diagnosis using genetic biomarkers. Genotyping the PNPLA3 I148M polymorphism has been suggested to help predict the severity and development of liver disease, particularly in children without obesity who are at risk of being overlooked [38].
This investigation clarified a process that was unrecognized to date by which obesity may cause liver steatosis. SREBP-1c could serve as a transcriptional target to regulate the steatotic effects of Sox4, and Sox4 expression was up-regulated in obese animals and patients with NAFLD. The HMG domain of SOX proteins is homologous to the HMG domain of the sex-determining region Y (SRY) protein, and 20 different Sox genes have been found in mammals. There may be context-dependent variation in SOX4’s biological functions and the genes it regulates downstream. Sox4 was discovered to control SREBP-1c expression, which can control glucose metabolism and insulin sensitivity. Sox4 was also shown to affect TG metabolism and Trb3 expression. Consequently, Sox4 may serve as a promising therapeutic target for the treatment of fatty liver disease and related metabolic syndrome [39].
Others have examined the link between obesity and three gene polymorphisms that cause liver steatosis in young people. In contrast to CK-18 levels, liver fat accumulation, triglycerides, and high very-low-density lipoprotein (VLDL) levels correlate with the rs1260326 mutation in the GCKR gene. CK-18 is related to two genetic variants: rs738409 in the PNPLA3 gene and rs2645424 in the FDFT1 gene. These findings provide credence to the concept that differences in genes that govern lipid metabolism play an essential role in the progression of fatty liver disease [2].
Despite increasing recognition, the diagnosis of NAFLD remains difficult in clinical practice, even though it is a potentially serious chronic liver disease. Genotyping for PNPLA3 might serve as an innovative noninvasive diagnostic method for elevated risk of developing fatty liver disease. Some variations of steatohepatitis are often referred to by acronyms such as NASH, ASH, BASH, CASH and DASH. The authors recommend identifying PNPLA3-associated steatohepatitis in patients with steatohepatitis who can become homozygous carriers of this risk allele and who are free of any other risk factors for fatty liver disease. The most recent studies looked at the link between PNPLA3 and liver health after a transplant. Independently of additional prostatic triggers, they discovered that homozygous carriers of the risk genotype had a 14-fold higher probability of developing graft steatosis [40].
However, no relationship was found between rs641738 in MBOAT7 and intra-hepatic fat content (HFF%) in their cohort of African-American children, and they identified an association between rs641738 and plasma high-sensitive C-reactive protein in the research cohort, namely PANIC. Despite these epidemiological analyses, the mechanisms linking this SNP to NAFLD have not been elucidated. Acyltransferase activity is provided by the protein produced by translating the MBOAT7 gene. It is difficult to identify why the impact of this gene mutation is only seen in Caucasians. However, it may be due to genetic differences in the metabolism of lipids. More research is required to validate these results and understand how this gene variation may contribute to NAFLD and insulin resistance in children. In a sample of Caucasian adolescents, researchers found that a specific genetic variant was linked to liver steatosis and insulin resistance. Caused by the link of genetic variation to NAFLD, insulin resistance was shown to be a common side effect of carrying the mutation. In overweight Caucasian children and adolescents, a particular variant of the MBOAT7 gene has been associated with insulin resistance and fatty liver [6].
A team found six genetic variations, including five missense variants and one intronic variant, all related to liver cT1 measured by MRI. In the UK Biobank, these variations are also associated with elevated levels of AST and ALT. Given the correlation between cT1 and histological fibrosis, the authors used cT1 as a continuous trait in their GWAS analysis. They compared it to the LIF score, a trilinear mapping of cT1 on a continuous scale from 0 to 4. Lower SLC39A8 expression levels have been associated with a missense mutation in SLC39A8 in the human liver, which has been predicted to be detrimental by the Polyphen-2 and SIFT programs. Manganese is a hepatotoxic metal ion, and ZIP8 controls its metabolism in the liver. Studies in mice show that SLC39A8 has a harmful function in liver inflammation and fibrosis. Necrosis, inflammation, fibrosis, and liver tumor growth are all caused by a lack of SLC39A8, which also disturbs the standard hepatocellular architecture. Novel correlations between polymorphisms in SLC30A10 and SLC39A8 and fibroinflammatory liver disease were found in a Mendelian randomization study. All kinds of biological activity are based on heavy metal cofactors, and these genes are essential for their delivery [9].
It is evident that proper fat oxidation suppresses liver fat production. However, excessive oxidation of fatty acids promotes oxidative stress, and FFA oxidation plays a complex role in the pathogenesis of NAFLD. The ‘gain of function’ polymorphisms in genes that encode proteins involved in peroxisomal and microsomal fats can predispose to NASH because these activities create ROS. NASH susceptibility may be increased by ‘loss-of-function’ mutations in genes involved in these processes. A variant in the IL-10 gene that leads to reduced promoter activity may play a role in the etiology of alcoholic liver disease (ALD). Patients with ALD tend to have more significant autoantibody titers. Major studies looking at the haplotype of the CTLA4 gene have not conclusively established that the Ala polymorphism in exon 1 of the cytotoxic T lymphocyte antigen 4 (CTLA-4) exon 1 is a susceptibility allele for ALD. Polymorphisms in genes encoding matrix metalloproteinase 3, PPAR-γ, TGF-β1, connective tissue growth factor (CTGF), and other fibrogenic adipocytokines can affect the inheritance of severe fatty liver disease. Future research must focus on identifying the specific genetic variables that predispose individuals to ALD and NAFLD. Large samples of both patients and healthy volunteers should be used in these investigations, as well as whole genome scans of single nucleotide polymorphisms and mutagenesis experiments in mice [41].
The causes of hyperhomocysteinemia in their research included aging, B vitamins, genetic abnormalities, polymorphisms of enzymes involved in homocysteine (Hcy) metabolism, and lifestyle variables, including smoking and inactivity. Insulin has been shown to affect the activity of metabolic enzymes involved in the turnover of Hcy in rat models and increased plasma. Hcy levels have been associated with insulin resistance and hyperinsulinemia in women with polycystic ovary syndrome, regardless of BMI. Patients with NAFLD had higher Hcy levels and a higher frequency of homozygote mutations MTHFR 677C/T and MTHFR 1298A/C than healthy controls; however, this may be due to selection bias in case-control studies using hospital controls [42].
Hepatocytes, HSCs, and endothelial cells express NOX4, although its involvement in NASH is unclear. There was no correlation between the NOX4 polymorphism and fibrosis in the liver. SNP-675 T/A in the CYBA gene was not linked to NAFLD, but is known to be functional and protects Type 1 diabetics from developing diabetic nephropathy. In GWAS, markers of steatosis, advanced fibrosis, and an increased risk of HCC were found to be associated with NAFLD at two different loci: PNPLA3 and TM6SF2. In a sample of people with NAFLD, the authors found that a mutation in the PNPLA3 gene was related to increased fibrosis [43].
Finally, it should be explicitly noted that genetic factors and their associations reviewed in the paper unfortunately cannot be used in clinical practice, either for diagnosis, for treatment, or for surveillance strategies.
Conclusions
Cirrhosis and liver failure may be the end result of NAFLD. Treatments exist to help people with the condition cope with its symptoms and have a better quality of life, but there is no cure. A thorough understanding of the signs and causes of the illness is essential for prompt diagnosis and therapy. However, more research is needed to make this study a reality, particularly with respect to early diagnosis and personalized therapy for pediatric and adolescent NAFLD. However, by compiling the existing literature, this study found compelling evidence for the existence of a genetic modifier that significantly affects the risk of NAFLD and the severity of the disease and its effects on the liver. In conclusion, patients with NAFLD with an uncommon allele may have more severe forms of FL and atherogenic dyslipidemia. The PNPLA3 variations found in the GWAS databases can be used to identify people at high risk for developing NAFLD. NAFLD may be exacerbated by a diet rich in saturated fat. It is possible that the stomach lining also functions as an endocrine gland. Obese people with biopsy-proven NAFLD had higher levels of NPY, NOX4, CYBA-675 T/A, metabolic syndrome, waist circumference, diet, and physical inactivity.





