
Introduction
Hypermobile Ehlers-Danlos syndrome (hEDS, OMIM#130020) is a non-inflammatory, autosomal dominant (for most cases) connective tissue disorder. According to the 2017 EDS International Classification, it is perhaps the most common hereditary connective tissue disorder with a prevalence estimated at 1/5000 [1]. Clinical manifestations include joint hypermobility with recurrent joint dislocations, chronic, widespread pain, mild hyperextensibility of the skin, recurrent or multiple abdominal hernias, atrophic scarring, gastrointestinal disorders, cardiovascular, immune, and nervous system dysfunctions, and in women a high-risk of premature delivery [2]. hEDS, unlike other types of EDS, has no known genetic aetiology, so diagnosis is conducted based on a person’s medical history, a physical examination, and exclusion of other types of EDS after genetic tests. Due to many clinical features that are common with other types of EDS (especially with the EDS classical type) and other non-inflammatory connective tissue disorders, proper recognition of this condition is difficult in many patients. Some reports point to the TNXB gene as an hEDS genetic factor [3, 4]. However, the fact that mutations in TNXB were detected only in a small percentage of hEDS as well as their recessive inheritance patterns suggests that TNXB is not the main hEDS-related gene. Investigations with the application of a next-generation sequencing (NGS) connective tissue panel, performed in hEDS patients, also do not identify disorder-associated genes. In several studies, new mutations were detected in well-known genes encoding extracellular matrix proteins, such as COL5A1, COL5A2, and COL3A1 [5–7].
In an investigation by Chiarelli et al., expression profiling of some ECM proteins in hEDSpatients’ fibroblasts was done. The study showed that ECM proteins, especially collagen type I, III, and V, as well as fibrillin and fibronectin, were detectable only in patients’ cell cytoplasm, whereas in intercellular spaces they were either not visible or only a few thin fibrils were present. Meanwhile, in healthy control cells, proteins were visible inside and outside the cells [8]. The cause of the dysfunction of ECM protein transport from cytoplasm to the intracellular space in hEDS patients is unknown. In the pathway of ECM protein transport, many molecules are involved in several stages. One of these proteins is heat-shock protein 47 (HSP47) encoded by the SERPINH1 gene.
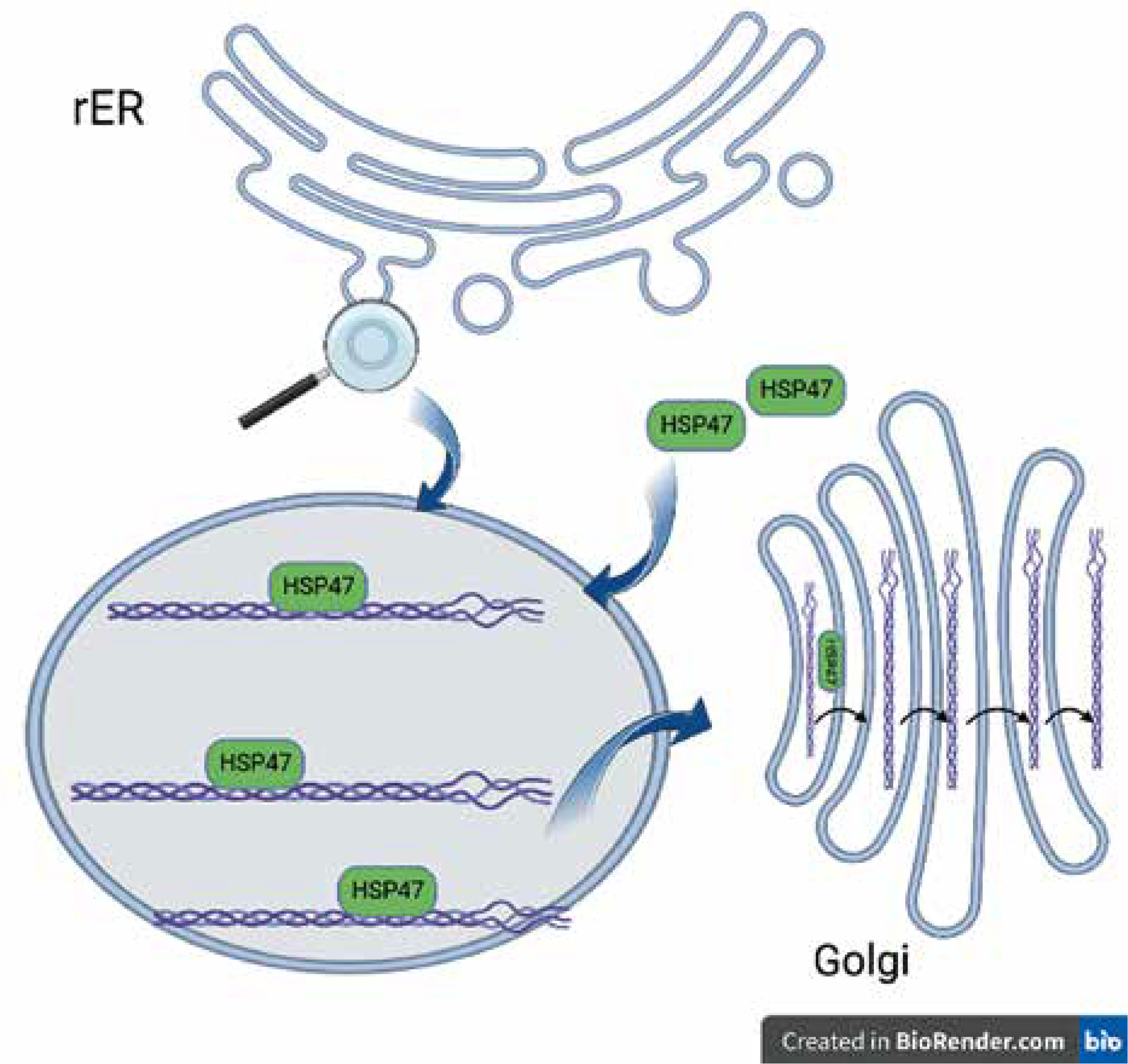
Collagen, the most abundant human protein, is a major component of the extracellular matrix (ECM) in tissues such as skin, bone, ligaments, tendons, arteries, and veins, as well as the gastrointestinal tract and respiratory system. Collagen biosynthesis is a complex, multistage process incorporating many molecules. It takes place in the rough endoplasmic reticulum (rER), and then the collagen molecule is transported via the Golgi apparatus to the extracellular space. All human proteins, including collagen molecules, require proper protein folding, maturation, and secretion processes. In the lumen of the rER collagen molecules undergo a series of posttranslational modifications, e.g. hydroxylation of propyl and lysyl residues by propyl 4-hydroxylases (P4H), propyl 3-hydroxylases (P3H), and lysyl hydroxylases (LH), respectively, and glycosylation of hydroxylysyl residues by glycosyltransferases. Other essential proteins involved in collagen folding and maturation are chaperone proteins such as HSP47 [9]. Heat-shock protein (HSP) 47 is a rough endoplasmic reticulum resident molecular chaperone, belonging to the serpin (serine protease inhibitor) superfamily [9, 10]. HSP47 is essential for the proper assembly of procollagen in the rER, which is eventually transported across the Golgi apparatus to the extracellular space (Figure 1). HSP47 was shown to directly bind to types I, II, III, IV, and V of collagen. It has been proposed that HSP47 serves 2 functions as a molecular chaperone in the collagen synthetic pathway: inhibition of local unfolding procollagen, and binding and stabilization of the triple-helical form of procollagen, and prevention of procollagen aggregation in the rER (prevention of rER-stress) [11, 12].
Figure 1
Collagen folding and HSP47. HSP47 binds to triple-helical procollagen in the rER and dissociates in the Golgi apparatus [9]. Figure created by BioRender
HSP47 is encoded by the SERPINH1 gene consisting of 5 exons. It has never been examined in hypermobile-type Ehlers-Danlos syndrome (hEDS) patients. The role of HSP47 has been evaluated in Osteogenesis Imperfecta (OI), but never in other connective tissue disorders. However, many hEDS patients share the phenotype with OI patients in whom the disorder develops based on COL1A1 variants [13–17]. Ishida et al. investigated biallelic knockout mice (HSP47-/-) cells for the role of HSP47 in type I collagen maturation and secretion from the rough endoplasmic reticulum to the Golgi apparatus. The secretion rate of this type of collagen was reduced in comparison to mouse cells HSP47+/+. They also noticed that in HSP47-/- cells, collagen molecules accumulated in the rER, compared to HSP47+/+ cells, in which such accumulation was not detected [18].
Aim
The aim of the present study was a sequencing analysis of the SERPINH1 gene and an evaluation of the potential role of variants of this gene on the aetiology of the hEDS.
Material and methods
The study group included 100 hEDS patients of Polish origin, women (84) and men (16), aged 17–63 years (median: 31 years). Patients were enrolled in the study by experienced clinical geneticists, according to the 2017 International Classification of the Ehlers-Danlos syndrome diagnostic criteria [1]. Joint hypermobility was evaluated on the Beighton scale. The control group consisted of 100 volunteers from the general Polish population matched by age and sex with the investigated group, who were healthy (including lack of EDS) at the time of the investigation and without a history of EDS in the family.
All hEDS patients or their parents provided informed consent to participate in the study. Consent to publish clinical/genetic data was also obtained from the patients or their parents.
The study was approved by the Ethics Committee of Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University in Torun, Poland (KB485/2013).
The analysis was performed on genomic DNA (gDNA), which was extracted from leukocytes (fibroblasts were not available) by QIAamp DNA Mini Kit (Qiagen, Germany) using standard procedures. In all patients, other types of EDS or other connective tissue disorders were excluded by testing them with NGS technology (Illumina NextSeq 550). The connective tissue disorder customer panel included COL5A1, COL5A2, COL3A1, COL1A1, COL1A2, TNXB, ADAMTS2, PLOD1, FKBP14, ZNF469, PRDM5, B4GALT7, B3GALT6, SLC39A13, CHST14, DSE, COL12A1, C1R, C1S, SEC23A, SEC24D, COL6A1, COL6A2, COL6A3, COL9A1, COL9A2, FBN1, FBN2, FLNA, and FLNB. Copy number variant (CNV) analysis was also performed. Sequencing data were aligned to the hg19 human reference genome. Based on the guidelines of the American College of Medical Genetics and Genomics, a minimum depth coverage of 30X was considered suitable for analysis. All patients negative for the NGS test were analysed for alterations in the SERPINH1 gene. Molecular analysis of the SERPINH1 gene was performed by Sanger sequencing (ABI3130XL) according to standard procedure (primer sequences available upon request). The pathogenicity of detected variants was assessed according to the ACMG guideline by Varsome [19, 20].
Results
All patients were tested by NGS multi-gene panel for connective tissue disorders, such as EDS, Osteogenesis Imperfecta, Marfan syndrome, and others. No pathogenic or likely pathogenic variants were detected in any of them. Copy number variants were also not detected.
The next step of the investigation was the sequencing of the SERPINH1 gene by Sanger sequencing.
Among 100 tested patients, 4 different types of missense variants (heterozygote) were detected. All SERPINH1 alterations were classified as benign according to ACMG guidelines (Table 1).
Table 1
Characterization of variants detected in the SERPINH1 gene
No pathogenic or probably pathogenic alterations were found. Variants were located in all exons of the gene, except for exon 1, which is non-coding. Part of exon 2 (from nucleotide number 60) to the beginning of exon 5 (nucleotide number 1159) correspond to the N-terminus part of the protein (Figure 2). All detected variants were found in the N-terminus protein domain, in C-terminus, no variant was located.
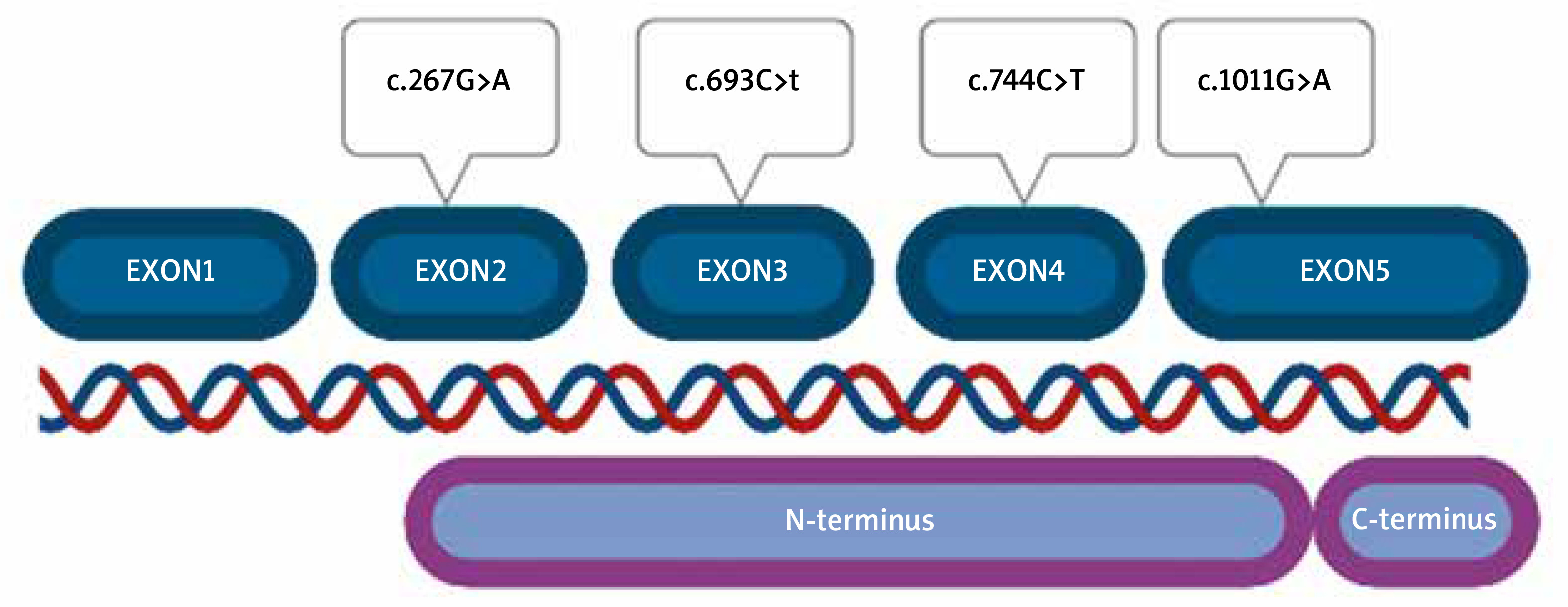
We also noticed that there were differences between the location of variants and patients’ clinical symptoms (severity and intensity of the symptoms, sex, age of onset of the disease), and also family history etc.
Discussion
Previous molecular analysis of the hypermobile type of Ehlers-Danlos syndrome focused mainly on a wide range of sequencing analyses of genes encoding connective tissue molecules. Despite intensive investigation, genes related to hEDS have not been identified. Some patients with EDS may share symptoms with patients with a mild form of Osteogenesis Imperfecta; furthermore, cases of EDS patients with COL1A1 mutations (a gene strictly connected with OI) are well known [15, 17, 21, 22]. Some phenotype-genotype similarities between EDS and OI were an implication for us to determine the role of another OI gene, SERPINH1, in the molecular background of hEDS [13]. Mutations in the SERPINH1 gene were described in a rare type of OI but have never been analysed in hypermobile Ehlers-Danlos syndrome. In our investigation among 100 hEDS patients, we did not identify pathogenic or probably pathogenic variants. Although only benign variants were detected, which play no role in the pathogenesis of hEDS, we should take into account mechanisms other than gene structure alterations, which may have an impact on collagen and other ECM proteins transport.
We are aware that the gene investigated by us encodes a protein that is only a part of the collagen/ECM secretion pathway, and the potential role of its disrupted distribution may be one of the mechanisms underlying hypermobile Ehlers-Danlos.







