
Hailey-Hailey disease (HHD, OMIM 169600) is a kind of autosomal dominant dermatosis. Hailey-Hailey disease is caused by mutations in the ATPase, Ca2+ transporting, type 2C, member 1 gene (ATP2C1) located at chromosome 3q22.1, encoding for human secretory pathway Ca2+/Mn2+ ATPase protein 1 (hSPCA1) which plays an important role in controlling Ca2+ concentration in the cytoplasm and Golgi apparatus of human keratinocytes [1]. Hailey-Hailey disease patients present with recurrent pruritic vesicles, painful erosions, and scaly erythematous plaques, which are caused by intraepidermal acantholysis due to retraction of keratin filaments from the desmosomal plaque and formation of perinuclear aggregates [2]. The disease most commonly affects intertriginous areas symmetrically such as the neck, axillae, groin, and perineum. Mechanical trauma, ultraviolet radiation, heat, sweating, or infection can cause exacerbations [3]. Here we report three Chinese families with HHD and identified three novel mutations of ATP2C1 sequence variations.
Three Chinese HHD families’ patients, unaffected family members, and 100 unrelated normal individuals were collected for mutation analysis. This study was previously approved by our local ethics committee, the Institutional Ethical Review Boards of the Peking Union Medical College and written informed consent was obtained from all subjects. The proband of family 1 was a 45-year-old woman with a 10-year history of HHD presenting with recurrent erythematous, erosive plaques with fissures. These lesions spread over bilateral axilla, groin and midriff area (Figure 1). Her mother and two sisters had similar lesions in their armpits and groin. The proband of family 2 was a 51-year-old man presenting with recurrent erythematous plaques, vesicles, erosions and crust involving the neck, bilateral axillary and inguinal areas with a 15-year history (Figure 2). The lesions were aggravated by sweating or hot weather. And the lesions flared up when he drank alcohol. His father and sister had similar lesions in their armpits and groin. The proband of family 3 was a 32-year-old man with a 4-year history of typical skin lesions of HHD (Figure 3). His mother had similar lesions in her neck, armpits and groin and midriff area. And all patients were confirmed by the histopathological test.
Figure 1
A – Clinical picture of the proband of family 1, B – pedigree of family 1, C – sequencing confirmed in the patients of family 1 (c.1241C>T)
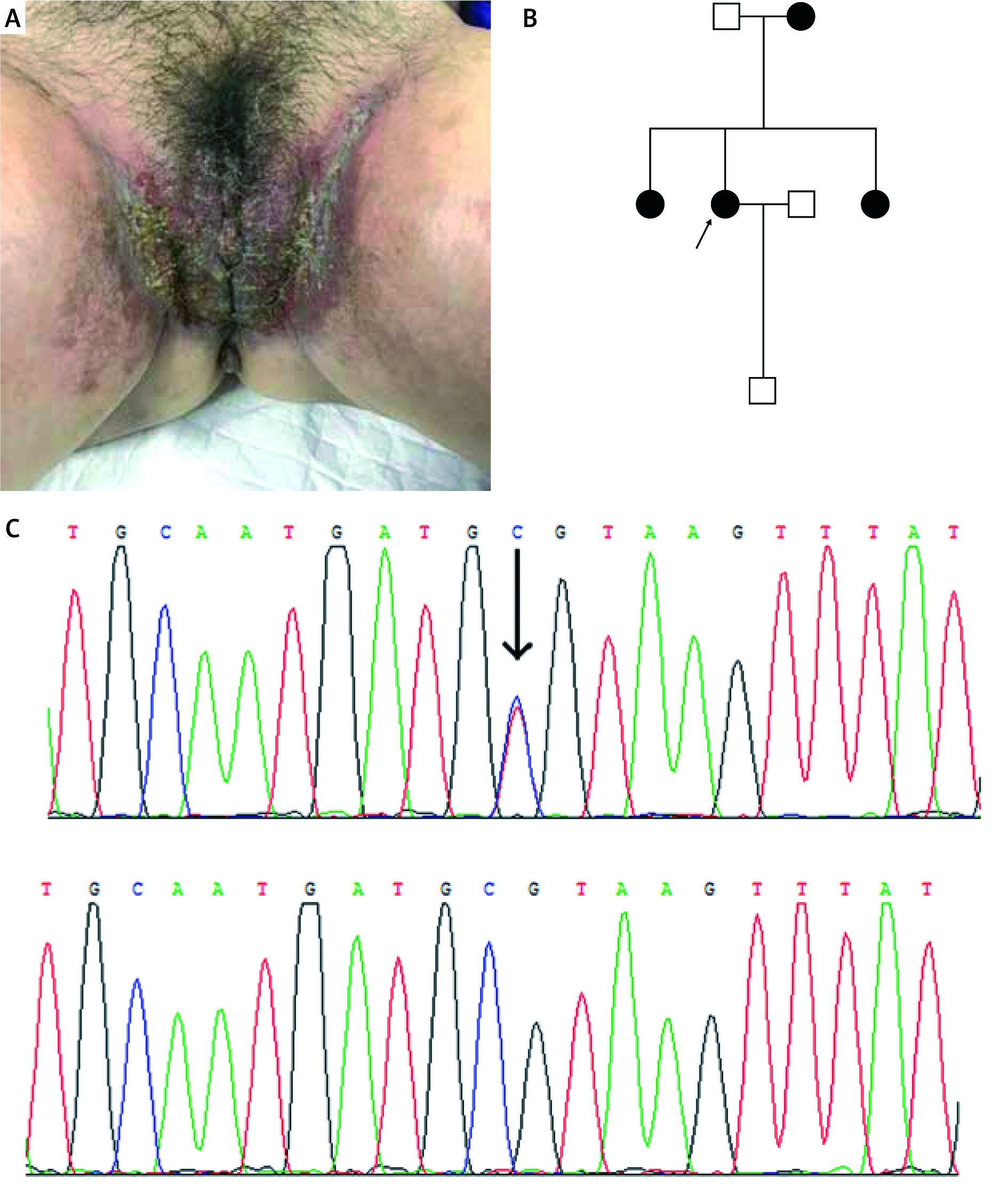
Figure 2
A – Clinical picture of the proband of family 2, B – pedigree of family 2, C – inverted sequencing confirmed in the patients of family 2 (c.473_474delGT)
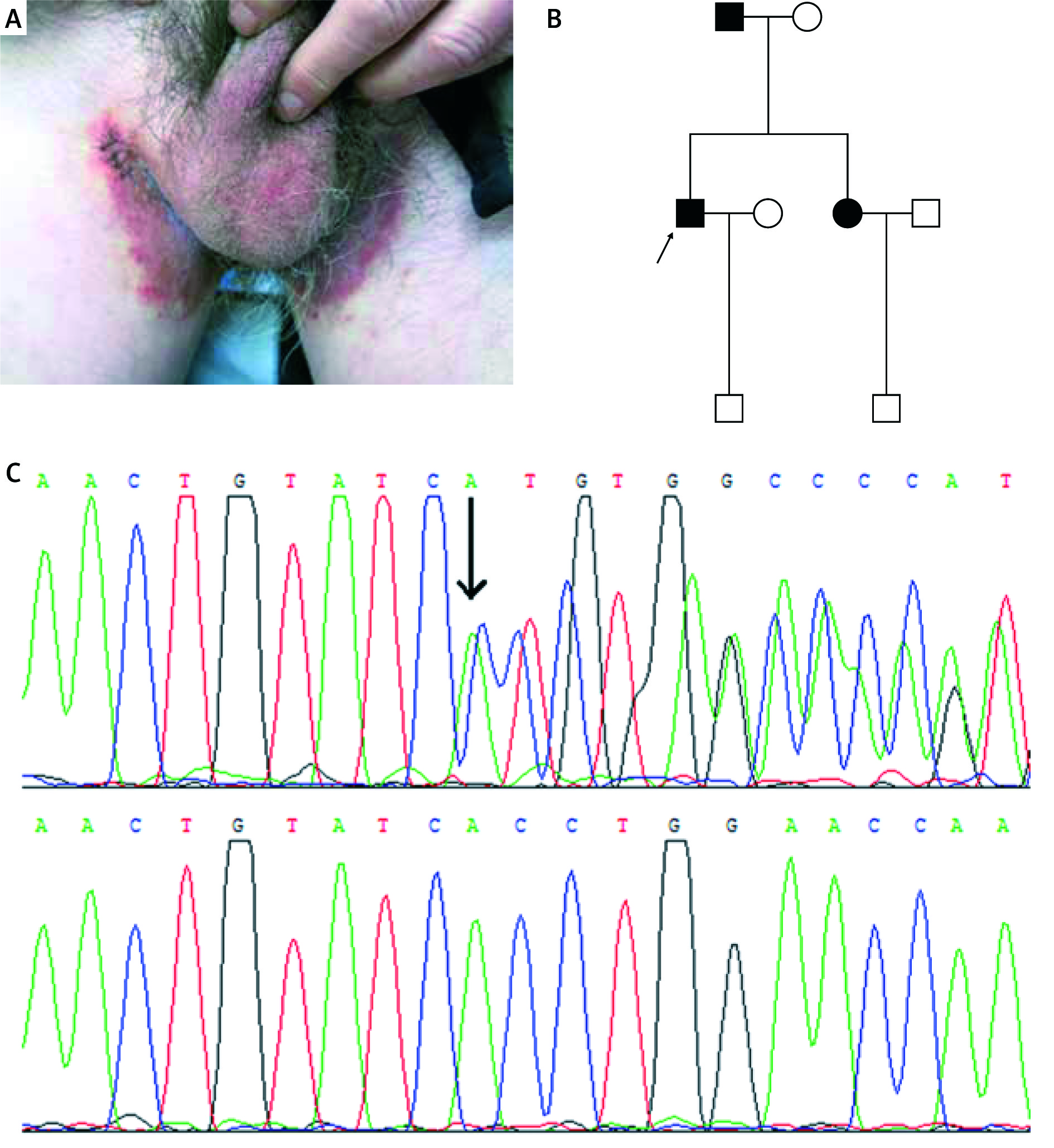
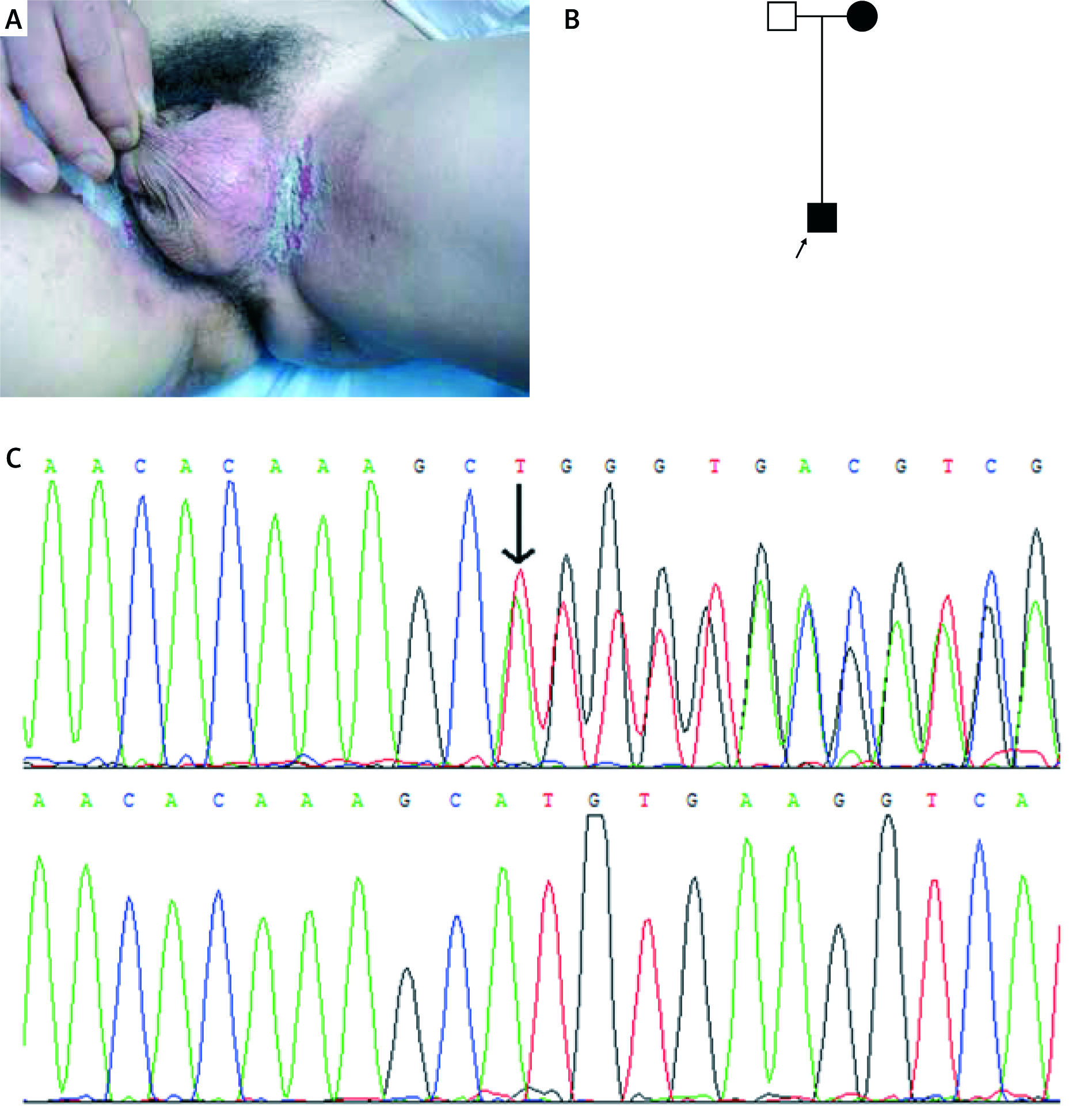
Figure 3
A – Clinical picture of the proband of family 3, B – pedigree of family 3, C – inverted sequencing confirmed in the patients of family 3 (c.2436_2440delCACATinsGGCACACA)
Genomic DNA samples were extracted from peripheral blood samples of HHD patients, unaffected family members, and 100 unrelated healthy individuals using the Wizard® Genomic DNA Purification Kit (Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s protocol. 27 pairs of primers flanking all exons of the ATP2C1 gene were designed using the web-based version of the Primer 3.0 program (http://www.genome.wi.mit.edu/cgibin/primer/primer3_www.cgi).
Primer sequences are available on request. Genomic DNA was amplified by polymerase chain reaction (PCR). After the amplification, the PCR products were purified using the QIA Quick PCR Purification Kit (Promega) and sequenced using ABI PRISM® 3730 automated sequencer (Thermo Fisher Scientific, Waltham, MA, USA.) Sequence comparisons and analyses were performed using Phred-Phrap-Consed v12.0 program (http://www.phrap.org).
Three heterozygous mutations of the ATP2C1 gene were identified in these pedigrees with HHD (Table 1). In family 1, missense mutation c.1241C>T (p.Ala414Val) in exon 15 was found in the proband and affected members, but not in unaffected members (Figure 1). In family 2, small deletion mutation c.473_474delGT (p.Asp159Tyr fs*8) in exon 7 was identified in the proband and affected members (Figure 2). In family 3, small indel mutation c.2436_2440delCACATinsGGCACACA (p.Phe812_Cys814delinsLeuAlaHisSer) in exon 25 was identified in the proband and affected members (Figure 3). In addition, these nucleotide substitutions were not found in 100 unrelated control individuals.
Table 1
Three novel mutations of ATP2C1 gene identified in this study
In our study, three heterozygous mutations of the ATP2C1 gene were identified in the three pedigrees with HHD. One missense mutation of c.1241C>T (p.Ala414Val) in exon 15, one small deletion mutation c.473_474delGT (p.Asp159Tyrfs*8) in exon 7, and one small indel mutation c.2436_2440delCACATinsGGCACACA (p.Phe812_Cys814delinsLeuAlaHisSer) in exon 25 were identified in three families, respectively (Table 1). All of the novel mutations were reported for the first time. The fact that none of these mutations was found in 100 unrelated control individuals indicates the mutations of the ATP2C1 are causative mutations rather than a rare polymorphism. Using the tool of PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), it is reported that these two mutations of c.473_474delGT and c.2436_2440delCACATinsGGCACACA are predicted to be probably damaging with a score of 1.000 (sensitivity: 0.00; specificity: 1.00), and the mutation of c.1241C>T is predicted to be probably damaging with a score of 0.998 (sensitivity: 0.27; specificity: 0.99). Tertiary interactions with other parts of the protein can be viewed as an environmental ‘buffer’ that helps diminish the effects of helix destabilizing mutations in helix 1, especially in the light of high tolerance of Ala to Val substitutions in helices. The missense mutation of c.1241C>T(p.Ala414Val) is invariant in all homologs except plasma membrane ATPases, and the residues involved are essential for ATP2C1 function. It raises the possibility that a decrease in Ca2+ concentration in the Golgi is caused by haploinsufficiency of ATP2C1.
The APT2C1 gene, located on chromosome 3q21.1, spans approximately 30kb and is composed of 28 exons [4]. A mutation of the ATP2C1 gene was first reported in patients with HHD in 2000 [5]. Since then, more than 150 mutations in the ATP2C1 gene have been reported, including nonsense, missense, frame-shift and splice-site mutations [6]. HSPCA1, which is encoded by the ATP2C1 gene, consists of an actuator domain, nucleotide-binding domain, phosphorylation domains, five stalk helices and 10 transmembrane helices [7]. While it is not quite clear how the disruption of ATP2C1 function results in the epidermal defects seen in the skin lesions. One possibility is that a decrease in Ca2+ concentration in the Golgi caused by haploinsufficiency of ATP2C1 could lead to a decrease in glycosylation or missorting of cell-cell adhesion molecules, such as desmosomal proteins. This may cause inability to maintain structurally intact desmosomes, leading to the acantholysis characteristic of HHD [8]. No clear genotype-phenotype correlation was found in HHD patients. Age of onset, severity or progression could not be attributed to the mutation location or type in the putative protein structure. Modifying genes and environmental factors may greatly influence clinical features of the disease.
In conclusion, we identified three novel heterozygous mutations in the ATP2C1 gene from three Chinese families with HHD. This study enriches the database of mutational analyses and helps extend our knowledge of HHD. Further research is required to clarify the genotype-phenotype correlation.







