
A 4-year-old boy from a nonconsanguineous Chinese Han family was referred to the Department of Dermatology, The East Division of The First Affiliated Hospital, Sun Yat-sen University, China, because of dyschromatosis. He was born at full term through an uncomplicated delivery, and developed photophobia as well as bilateral lop ears at the age of 3 months. When he was 6 months old, he developed hypohidrosis that was over the whole body except the neck and outer aspects of the thighs, he also had mild fever of about 37.5°C during the summer or after physical activity, leading to erythema. Hereafter, generalized hyperpigmentation with mottled hypopigmentation developed over his whole body. He had normal growth and normal mental development, and there were no systemic manifestations as well as associations such as pruritus, pain, or burning sensations since his birth. His mother, and grandmother as well as the maternal grandmother on mother's side had multiple, asymptomatic, brownish macules in linear and whorled patterns over the trunk, axillae, groin and extremities after birth, which relieved after adolescence. His elder sister had cleft palate in addition to linear hyperpigmentation. During a 4-year follow-up, the patient’s hypohidrosis and the rise of body temperature had mild improvement although no treatments were ordered, while mottled hypopigmentation increased and was accompanied by more severe hyperpigmentation.
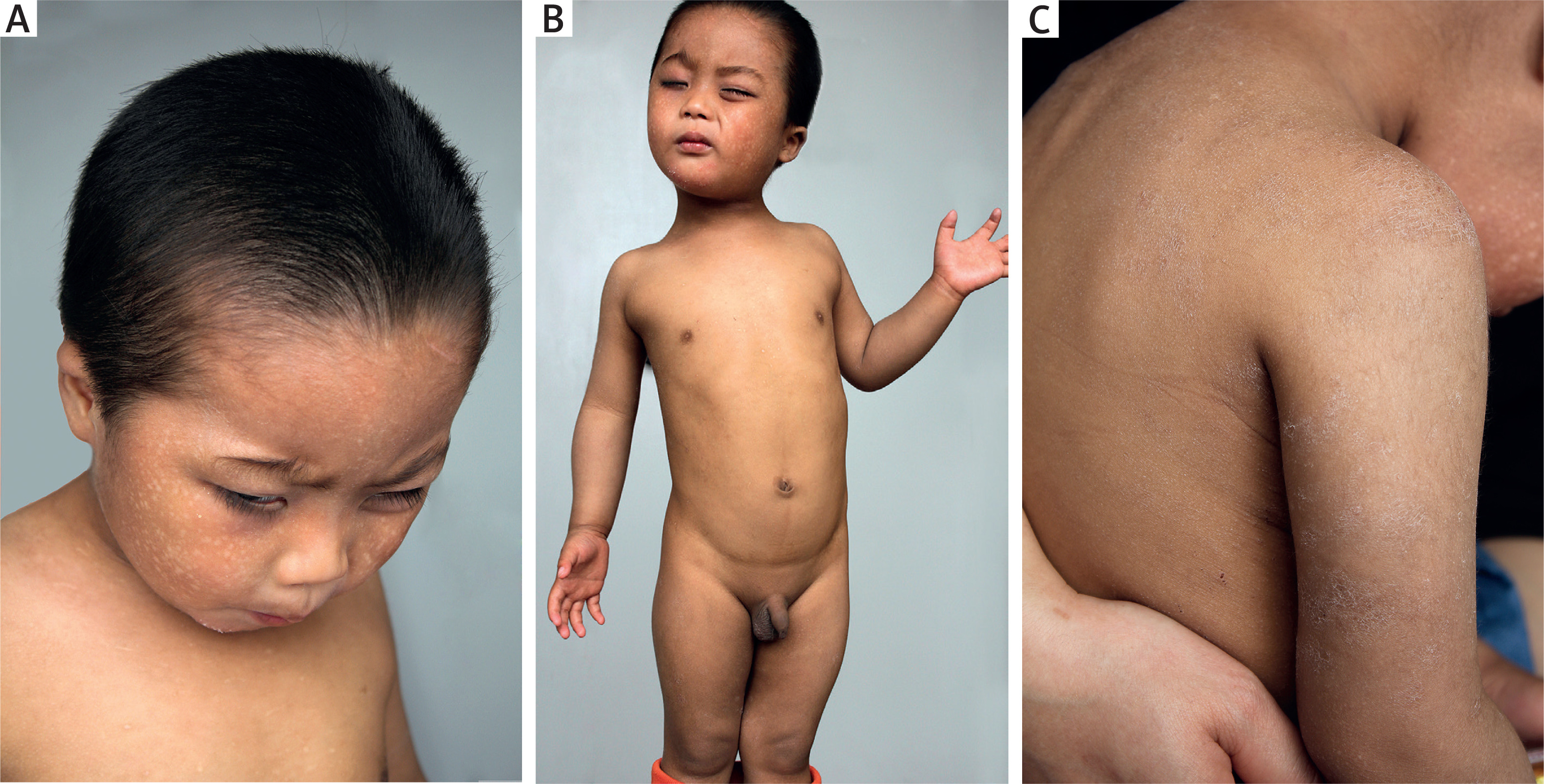
Cutaneous examination showed diffuse reticulate hyperpigmentation with mottled hypopigmentation over the whole body, associated with photophobia, coarse and dried skin, upswept frontal hairline and flared eyebrows (Figures 1 A–C). Skin biopsy was refused.
Figure 1
Diffuse reticulate hyperpigmentation with mottled hypopigmentation on the whole body as well as upswept frontal hairline, flared eyebrows and photophobia (A, B), and coarse and dried skin (C)
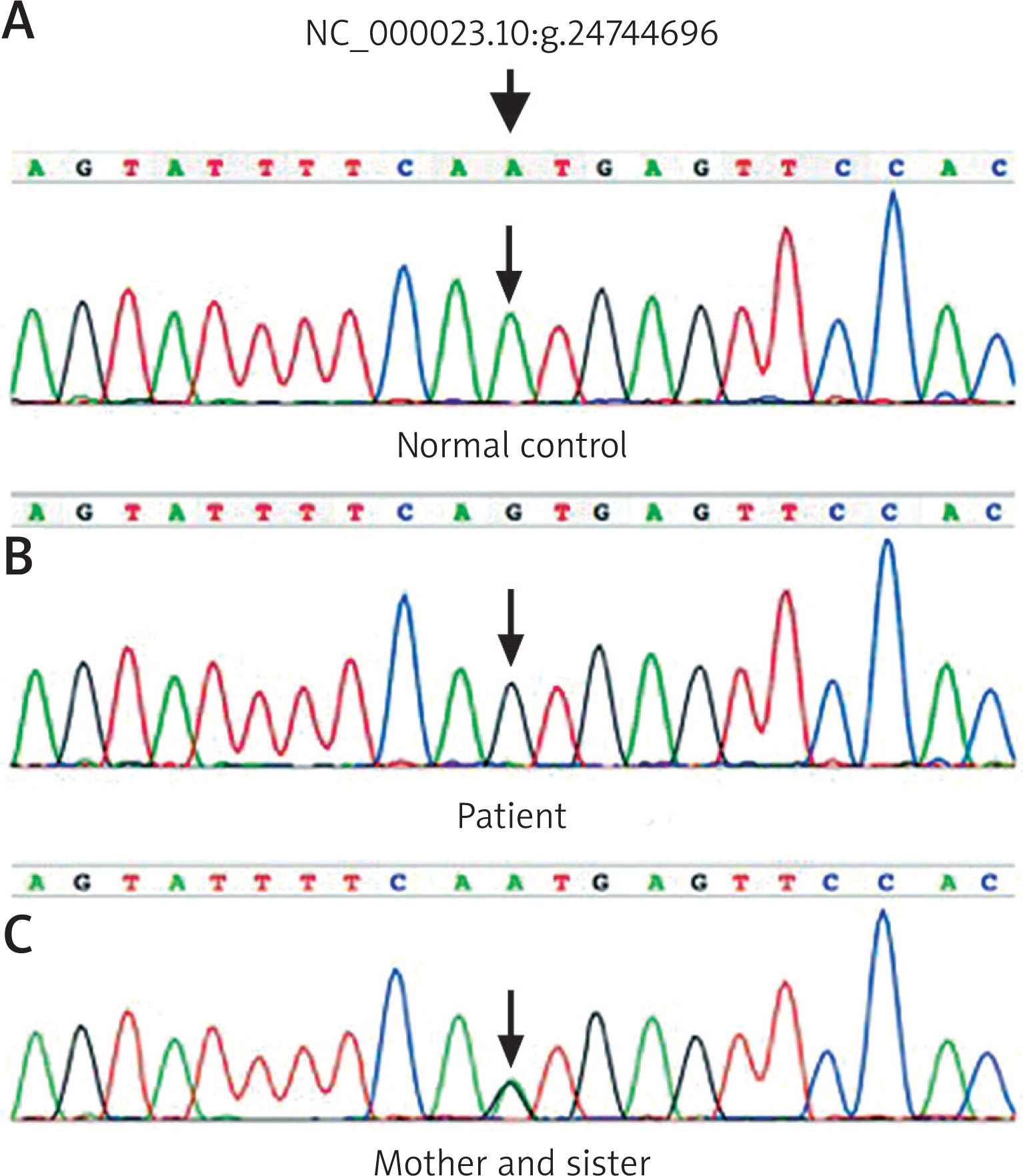
After informed written consent was obtained from the patient’s parents and other family members, DNA was extracted from peripheral blood of the patient and his family members according to standard methods. Then exome sequencing of the POLA1 gene was performed and revealed hemizygous mutation of NC_000023.10:g.24744696A>G for the patient and heterozygote for his mother and sister (Figures 2 A–C). Then a diagnosis of X-linked reticulate pigmentary disorder was confirmed.
Figure 2
Exome sequencing of the POLA1 gene shows normal control gene (A), hemizygous mutation of NC_000023.10:g.24744696A>G in the patient (B) and heterozygous mutation in his mother and his sister (C)
X-linked reticulate pigmentary disorder [XLPDR; MIM301220], first described in a Canadian pedigree [1], is an exceptionally rare genodermatosis inherited as X-linked recessive trait [1–9]. In males, it is always characterized by diffused reticulate dyschromatosis with photophobia, dry skin and typical facial appearance featuring upswept frontal hairline and flared eyebrows. Hypohidrosis is also a distinctive feature, with predilection for trunk and extremities [2, 3] as our patient presented. The systemic manifestations include recurrent infections and autoimmune reactions against various organs including respiratory, gastrointestinal, and neurological systems [2], however, not all patients including the present one had such presentations. The reasons for the absence of systemic manifestations remain unknown which needs further study. Males always have more severe symptoms than female carriers. Whereas the females always present patchy hyperpigmentation along the Blaschko’s lines alone that are similar to stage III incontinentia pigmenti and will alleviate and even disappear after puberty, but without systemic involvements [2, 4, 5], as did in our patient’s mother and his sister. Although cutaneous symptoms and peculiar facial appearance are considered to be the distinctive features of XLPDR [3, 4], absence of skin lesions in infancy had also been reported [4]; in rare instances, the reticulate hyperpigmentation developed until the patient was 8 years old [6]. The absence of skin lesions in early stage may lead to a delayed diagnosis for the patient. Up to date, 25 cases from different families including the present one have been reported [1–9]. However, it is not until 2015 that the mutation of c.1375-354A>G or g.24744696A>G in POLA1 gene was demonstrated as a causal factor for XLPDR [7], and all the patients with genetic demonstration including the present one were detected to have the same intronic mutation in POLA1. Because of the rarity of the disease, it remains to be elucidated whether XLPDR is caused by this mutation specifically, or this mutation stands for one mutation hotspot with additional mutations in POLA1 underlying XLPDR. Nevertheless, it is undoubtedly cost-effective to screen preferentially for this mutation in cases of XLPDR.
The exact mechanisms for XLPDR as well as POLA1 remain fully unknown. POLA1 protein, or DNA polymerase alpha catalytic subunit, is necessary for initiation of DNA replication and the synthesis of cytosolic RNA:DNA, which directly modulates interferon-regulatory factors [3, 7]. The reduction of POLA1 expression may result in diminished cytosolic RNA:DNA hybrids [3, 7], which are responsible for the negative regulation of interferon-regulatory factors, leading to increase of type I interferon [3, 7]. It has been reported recently that XLPDR was associated with a decreased number and selective cytotoxicity defect of NK cells [3]; this might be why the patients have autoinflammation and recurrent infection [6, 7]. Interestingly, no recurrent cutaneous infection has been described up to date. The histopathological features of XLPDR are similar in both sexes, including mild hyperkeratosis, acanthosis, basal hyperpigmentation, and pigmentary incontinence in the upper dermis [2, 4, 6]. Deposition of amyloid-like material in the papillary dermis and mild superficial lymphocytic perivascular infiltrate may also be present [2, 4, 6]. Direct immunofluorescence may reveal granular deposits of C3 at the dermoepidermal junction [6]. However, these histopathological findings are non-specific for the disease [2, 4]. The mechanisms for pigmentation also remain elusive. Accumulation of melanophages and amyloid-like materials had been found in the upper dermis [1, 2, 4, 5, 6], suggesting that those might be responsible for the hyperpigmentation. Of course, further studies are still required to verify this speculation and clarify the function of POLA1 in skin.
Differential diagnosis of XLPDR includes incontinentia pigmenti, primary cutaneous amyloidosis, Rothmund-Thomson syndrome, Kindler syndrome, congenital dyskeratosis, and Naegeli–Franceschetti syndrome [1, 4, 5, 8]. Based on the featuring presentations and inherited trait, it is not hard to make a correct diagnosis, sometimes, sequencing of the POLA1 gene is an optimal option. No therapeutics are recommended for the disease at present yet.







