
Introduction
Breast cancer (BC) is the most common type of malignancy affecting females worldwide, with about 2.1 million new diagnoses worldwide and 626,679 deaths estimated each year. The incidence of this malignancy has increased by about 20% over the past five years. It is also the main cause of mortality among females, affecting one in four women suffering from malignancy [1]. miRNAs are short non-coding RNAs that regulate gene output at the post-transcriptional level by targeting degenerate elements primarily in 3’untranslated regions (3’UTRs) of mRNAs and are involved in many biological processes, such as cell proliferation, cell cycle regulation, differentiation, apoptosis, and development. Individual miRNAs can regulate networks of hundreds of genes. Therefore, their dysregulation can lead to a variety of diseases including cancer. Individual miRNAs can regulate networks of hundreds of genes, but for the majority of miRNAs few, if any, targets are known. Misexpression of miRNAs is also a major contributor to cancer progression, thus there is a critical need to validate miRNA targets in high throughput to understand the contribution of miRNAs to tumourigenesis. In human BC, it has been shown that they can act either as tumour suppressors or oncogenes [2]. In particular, miR-10b was proven to play a prominent role in regulation of tumour invasion and metastasis. This miRNA expressed in metastatic BC cell lines is able to generate metastases when growing as a primary tumour in mice [3]. miR-335 was demonstrated and reported to play important roles in tumour development, including roles in tumour cell proliferation, survival, migration, invasion, and colonisation. Furthermore, it has been shown that this miR acts as a BC suppressor. However, the mechanisms by which miR-335 affects these biological processes in BC remain unclear [4].
miR-21 is significantly elevated in the majority of human tumours. It plays an important role in tumour formation and development, and it has been shown to be a key regulator of oncogenic processes. Iorio et al. reported that miR-21 is overexpressed in BC tissue and may be a useful marker of BC [5].
Two large classes of regulatory molecules working post-transcriptionally are microRNAs (miRNAs) and RNA-binding proteins (RBPs). Although various mechanisms are responsible for the dysregulation of miRNAs in cancer, the miRNA biogenesis pathway is almost controlled by cancer cells to promote dysregulated expression. Furthermore, many human miRNA genes are located at fragile sites or in other genomic regions that are subjects of mutation, deletion, amplification, or translocation in cancer [6, 7]. Recently, cancer regulation by miRNAs had been extensively studied and understood to such an extent that several miRNAs are nowadays classified as “oncomiRs” or, conversely, as tumour suppressor miRNAs [8]. As for miRNAs, the involvement of some RBPs in oncogenesis and tumour progression has been shown in several types of cancer [9]. Depending on the protein, the mRNA, and the biological context, the regular actions of RBPs may be positive (activators) or negative (suppressors) [10].
Positive actions in miRNA biogenesis result in them behaving as general enhancers of miRNA functions. Thus, this protein dysregulation is often observed in numerous types of human tumour cells. For example, overexpression of DICER1 in acute myeloid leukaemia cells [11], downregulation of miRNA biogenesis components in hepatocellular carcinoma [12], and DICER downregulation are used to predict poor prognosis in chronic lymphocytic leukaemia [13] and many other types of cancer [14].
In the second class of RBPs, which contrasts with miRNA function, the activity of miRNAs is hindered by RBPs, as shown in the study of p53 inducing the RBP RBM38, which limits the accessibility of miRNA sites on 3’UTRs of its target gene transcripts. Hence, an important proportion of wild type p53 tumours, like breast and prostate cancer, suggest an active mechanism to silence RBM38 in those tumours because it possesses DNA methylation of the RBM’s promoter region [15].
The functional relationship between miRNAs and RBPs can also be carried out via the regulation of RBP expression by specific miRNAs. Obviously, it results in a modulation of the effects achieved by RBPs and RBP-controlled genes, and this may be considered as an additional way for the miRNAs to control gene expression and then to contribute to the diversity of cancer cell characterisation.
Furthermore, due to the chemical nature of ribonucleic acid, miRNAs are regulated by RBPs which have an obvious effect on miRNAs precursor pathways in both nuclear and cytoplasmic processes in all cell types, DROSHA and DICER [16]. In defined conditions such as cancer, some other RBPs act specifically on selected miRNAs.
Also in this study we investigate interactions between miRNAs and other proteins that play main and important role in the cell cycle, and some are associated with BC. Among those proteins and genes, we cite: BRCA1 – since its cloning in 1994, numerous studies have established its role in diverse cellular and biochemical processes, such as DNA damage repair, cell cycle control, and transcriptional regulation as well as ubiquitination. BRCA is also well-known as a tumour suppressor implicated in familial breast and ovarian cancer [17]. In vivo, BRCA1 exists as a heterodimer with the BARD1tumour suppressor, and both proteins harbour a phosphate-binding BRCT domain. Thereby, the main role of BARD1 is an essential contribution to the tumour suppression activity of the BRCA1/BARD1 heterodimer [18]. Furthermore, a study by Ganesan et al. suggests that BRCA1 and BARD may form direct or indirect interactions with Xi-specific transcript (XIST) RNA, and they demonstrated that loss of BRCA1 can lead to a specific disruption of several characteristic features of inactive X chromosome (Xi), including the normal localisation of XIST RNA, and that this interaction may be part of a more general role of BRCA1 in the regulation of chromatin structure [19].
CHEK2 plays a key role in the cellular response to DNA damage, chromosomal stability, and regulation of mitosis. CHEK2 mutation along with a family history of BC imparts a 28–37% lifetime risk of breast carcinoma [20]. PTEN is a crucial tumour-suppressor gene, which may negatively regulate cell proliferation, renewal, and differentiation [21]. This gene had been shown to be dysregulated in many malignant tumours, especially BC [22]. Furthermore, down the expression of PTEN is associated with several outcomes in many diseases [23]. Computational tools allow us to understand better the mechanism of our bodies and accelerate research and innovation. Thus, tertiary structure analysis of miRNA–target interactions provides a more detailed understanding of siRNA/miRNA function, which could open new avenues for developing more accurate algorithms for target prediction. Current computational methods to identify mRNA targets of miRNAs use rules based on primary and secondary structure information. Furthermore, hidden Markov and Bayesian models coupled to Monte Carlo methods provide the possibility to get more information about the statements and the relations into complex systems using a stochastic model.
Here, in this study, the proteins and genes were selected from the literature as important targets, like miRNA target site prediction and miRNA-mRNA duplex prediction, and molecular docking between a variety of 40 proteins and three miRNAs: miR-10b, miR-21, and miR-335, were conducted to elucidate the protein-assisted molecular interaction between miRNAs and these proteins computationally.
Material and methods
The workflow of the method used in this study is described i n Figure 1.
Fig. 1
Workflow of the analysis. We used computational methods to predict the possible interactions between miRNAs and proteins. The first method was based on docking and chemoinformatics approaches to reveal the strengths and natures of the bonds based on docking 3D structures. The second method was based on HMM applied to amino acids and nucleotide sequences revealing essentially the positioning of the bonds. Afterwards, we proceeded to the comparison of the results obtained from the two methods
Data set
The sequences of miR-10b, miR-335, and miR-21 were collected from the miRBase microRNA database [27]. The crystal structures of proteins were collected from the PDB database; their accession numbers are shown in Table 1. All the ligands and antibodies were removed from the PDB structure files as well as the water molecules.
Table 1
Binding energy and RMSD values between proteins and miR-10b, miR-335, and miR-21
2D and 3D structures prediction
To predict 2D structures of miR-10b, miR-335, and miR-21 we used RNA structure software [28] and ViennaRNA Web Services [29]. However, the prediction of the 3D structure was made using the software RNAComposer [30] and SimRNAweb [31]. Evaluations and benchmarks were obtained based on the ModeRNA server [32].
Interaction protein-miRNA
Many studies have developed the interactions between miRNA, mRNA, and promoter genes. However, to our knowledge, and because during our search of the database no studies demonstrated direct interactions between miRNAs and our target proteins, 3D crystal structure is not available in databases. In this context, we used chemoinformatics tools with the objective to study the possible interactions between proteins and miRNAs, based on molecular modelling using docking protein-RNA and matching protein-RNA.
After pre-docking preparation, the miRNAs and protein 3D structures were treated by NP Dock HADDOCK 3.2 [33] and Hex 8.0 to predict the eventual interactions using docking protein-RNA tools with default parameters. The docking process was repeated four times for each protein-miRNA couple, and the average of the free binding energy was calculated, taking into consideration the number of bonds revealed by Discovery Studio 4.0.
Hidden Markov model Bayesian protein-RNA
In order to confirm our results of docking we used the HMM prediction method to compare its results with the docking results. Thus, in the present study, we used the aPPRove algorithm based on HMM [34] to validate the results using the HMM coupled to Bayesian models. The transition matrix was defined in order to study the transition probability among states. Whereas, for the probability from state a to b, the transition probability of leaving the state was defined according to the formula:
where h and g are set as pseudo-counts used in order to determine A and T probabilities. We defined g (i, j) for all possibilities of i and j, where i and j state in the pair hidden Markov model. The Viterbi algorithm was used in order to validate the scores of transmission and emission from one state to another.
Results and discussion
2D and 3D structures
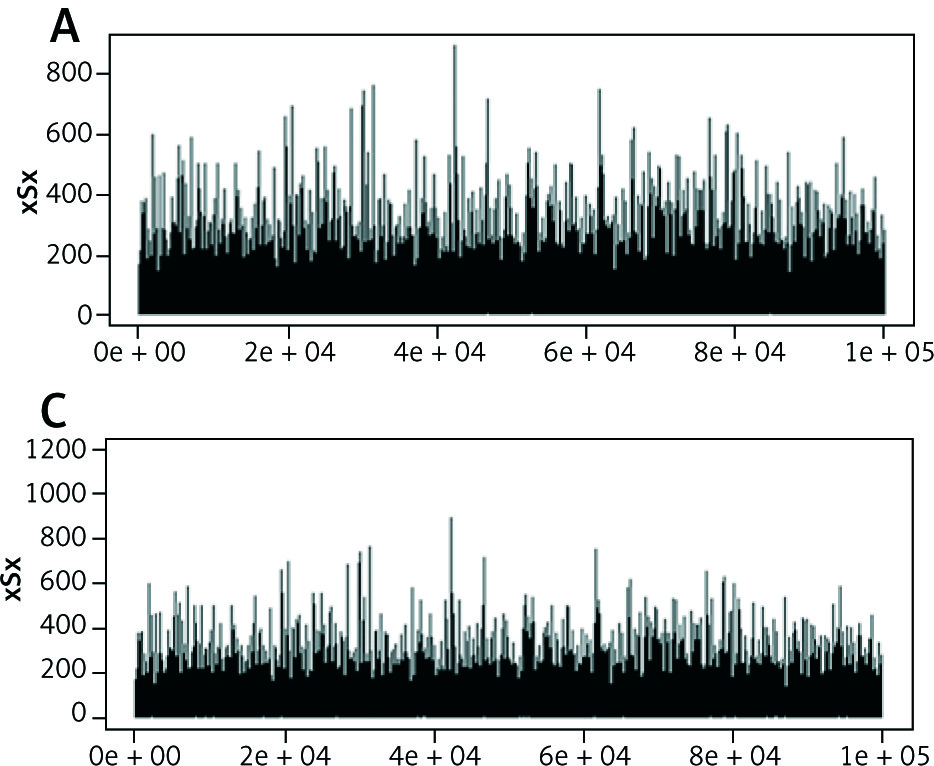
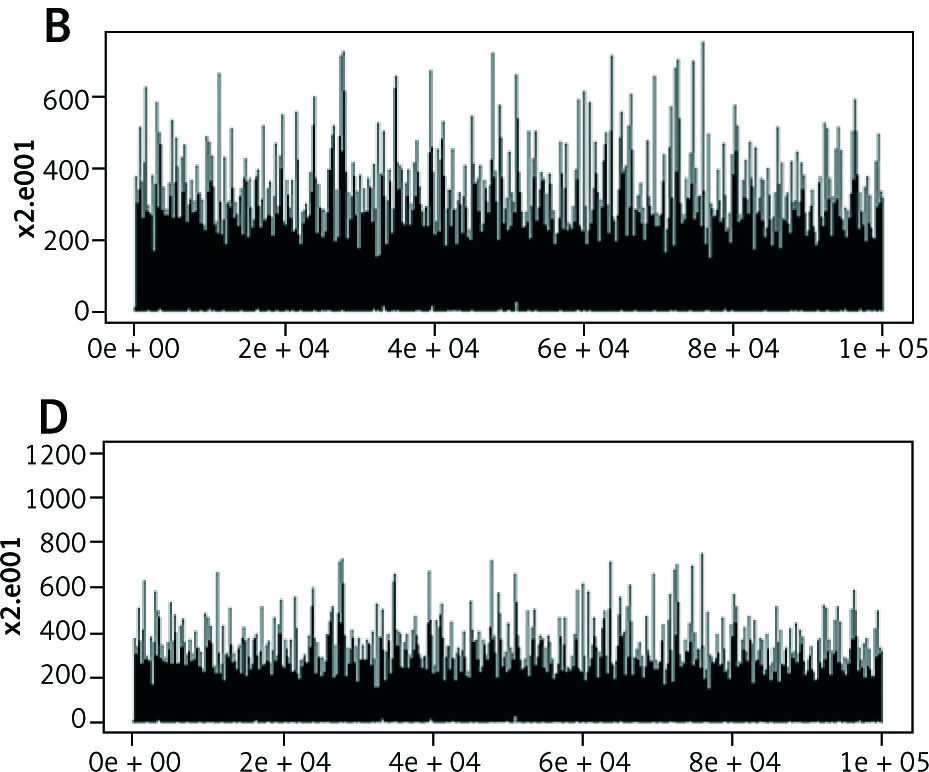
Tertiary structure-based computational methods are required for a more detailed analysis of the structural mechanisms of miRNA target recognition and post-transcriptional regulation. We used Monte Carlo (MC) methods, which are frequently applied to sample putative interaction geometries of proteins, including also possible conformational changes of the binding partners. Figure 2 demonstrates the score of prediction using HMM of amino acid interactions.
Docking and protein interactions
In order to study eventual interactions between miRNAs and proteins or genes, molecular docking with default parameters was carried out. Thereby, we tested by docking the possible interactions between our miRNAs and BRCA1, PTEN, BRCA1/BARD1, BRCT, CHEK2, BP1 BRCT7/8-BACH1, PI3K-g cancer, BRCA1/BRCT, E-cadherin, Mre11, HER2-, CDK1/CKS1, E1, E2, and E3 enzymes and ATM. As the literature has reported, a pocket of miRNAs implicates strong binding interactions at the molecular level between AGO protein and various miRNAs. Our work was mainly focused on the hypothesis that miRNAs can make covalent and non-covalent linkages with oncoproteins and suppressor proteins, which is not yet demonstrated by studies in vivo. According to the modelling of structures, we used chemical modelling tools, namely covalent and non-covalent docking and mathematical modelling by HMM, to illustrate and test this hypothesis. In general, miR10, miR335, and miR21 demonstrated their ability to create linkages with 3D protein structures, of which the long RNA or reference DNA sequences such as TATA box do not interact with the current protein. Thus, our study shows that miR-10b produces non-covalent bonds with allosteric sites of complex BRCA1/BARD1, which directly leads to an increase in or inhibition of their activity. Furthermore, we found that the possibility of interaction is favourable between miR-10b and oncoprotein E6, P27(KIP1)/cyclin A/CDK2 complex, Mre11 nuclease, which is essential for dsDNA break repair, Skp1-Skp2-Ck1, which plays a major role as mitotic regulatory protein, E-cadherin, which regulates cell growth, and BRCA1. The positioning of the interactions between the proteins and miR10 are located in different sites of the proteins according to the nature of the protein (Fig. 3).
Fig. 3
Interaction between miR-10b and A) E6, B) P27 (KIP1)/cyclin A/CDK2 com, C) Mre11, D) Skp1-Skp2-Cks1, E) E-cadherin, and F) BRCA1
On the other hand, we noticed that the strength of interactions was variable for each protein. Thereby, Table 1 shows the energy of binding of some studied proteins and target miRNAs. The interactions are significantly higher than for other proteins. We also found that these interactions are mostly covalent, with the presence of the non-covalent bonds enhancing the typical energy voices for stabilisation of complexes. It should be noticed that some proteins such as HE2 do not interact with the large RNAs and reference DNA. However, in the case of miR-10b and 335, these strong interactions elucidate the idea that the overexpression of miRNAs is not limited to the level of mRNA but also to the proteomic level, which explains and provides another pathway of research.
Furthermore, studies focused on expression profiling have shown that the dysregulation of miRNA expression has various origins, which provide these biomarkers with their tissue-specific characteristics. Growing evidence suggests that miRNAs can be regulated extensively at the levels of promoter transcription methylation, miRNA processing, RNA editing, and miRNA-target interactions [24]. Thereby, transcriptional regulation by nuclear receptors is the primary level of control for miRNA expression. This regulation can be achieved via three mechanisms: direct binding to the promoter regions of miRNAs, indirect regulation of miRNA expression through NR target genes, and involvement in the regulation of miRNA biogenesis [25]. BRCA1 regulates the expression of miRNAs, which may, in turn, regulate the expression of BRCA1. MiRNAs are known to down-regulate the expression of the protein in two different ways: by inhibiting the expression or degrading mRNA [26]. This mechanism can have the same effect in dysregulation of BRCA1 function. These direct interactions allow us to conclude that the influence levels of miRNAs are not only genomic but can reach proteomic levels. Such direct interaction between miRNA and protein can influence the direct regulation of the action mechanisms in the biological system. The strength of the interactions between the crystallised structures and the miRNAs is very large compared to those observed with the same crystal structures and the DNA fragments obtained during the crystallisation; essentially those located at the promoter regions.
In conclusion, miRNAs play an increasingly conspicuous role in the regulation of epigenetic and post-transcriptomic effects in cancerogenesis and oncological processes. miR-10b binds with oncogenic genes, explaining the increased expression of miR-10b. The results demonstrate covalent linkages between miR-335 and P53 and other suppressor proteins in the activating regions, whereas the interaction with the oncogenic progeny may be less likely. On the other hand, interactions with viral-oncogenic and oncogenic proteins are observed at the RNA-protein docking level. This may constitute a new pathway to other hypotheses of degradation and regulation of tumourigenesis mechanisms.






