
Congenital lobar emphysema (CLE) is an innate malformation of the lungs, diagnosed in 1 in 20,000 live births [1]. It was described for the first time by Nelson in 1932, and later by Robertson and James in 1951 [1]. Erroneous development of the respiratory system in the third week of gestation leads to malformation of the lungs [1]. It is rarely diagnosed prenatally with the help of ultrasonography (USG) [2]. The pathology is due to partial obstruction of the bronchus causing hyperinflation of the pulmonary lobe [1, 2]. Most commonly absent or hypoplastic cartilage leads to the collapse of the bronchus, and during expiration to trapping of air [2]. Causes of acquired CLE are aspiration of the meconium, plaques of mucous, hypertrophy of the mucous membrane, aspiration of a foreign body, bronchogenic cyst and mediastinal tumor [1, 2]. Rare abnormalities of the vessels, e.g. abnormal pulmonary venous return, may also be the cause of CLE [2, 3]. According to some authors CLE can be hereditary [4]. CLE is accompanied by heart defects in 10% of cases, so all patients should have echocardiography diagnostics [1–4]. Other possible concomitant malformations are double superior vena cava and horseshoe kidney [1–4]. Most cases are diagnosed in neonates, with male preponderance (boy to girl ratio 3 : 1) [1–4]. Typically the left upper lobe is involved (43%), then the right middle lobe (32%) and the right upper lobe (21%) [3, 4]. CLE localized in the lower lobes is rather unique (2%) [3, 4]. Bilateral localization was also observed [1, 2]. Histological examination reveals alveolar overgrowth and normal structure of the pulmonary acinus [1–3].
CLE may be the cause of respiratory distress and cardiac arrest. Children with severe symptoms are treated with thoracotomy and lobectomy [1, 4]. It is postulated that asymptomatic CLE should be treated nonoperatively, because infections and malign malformations are rare [1, 4].
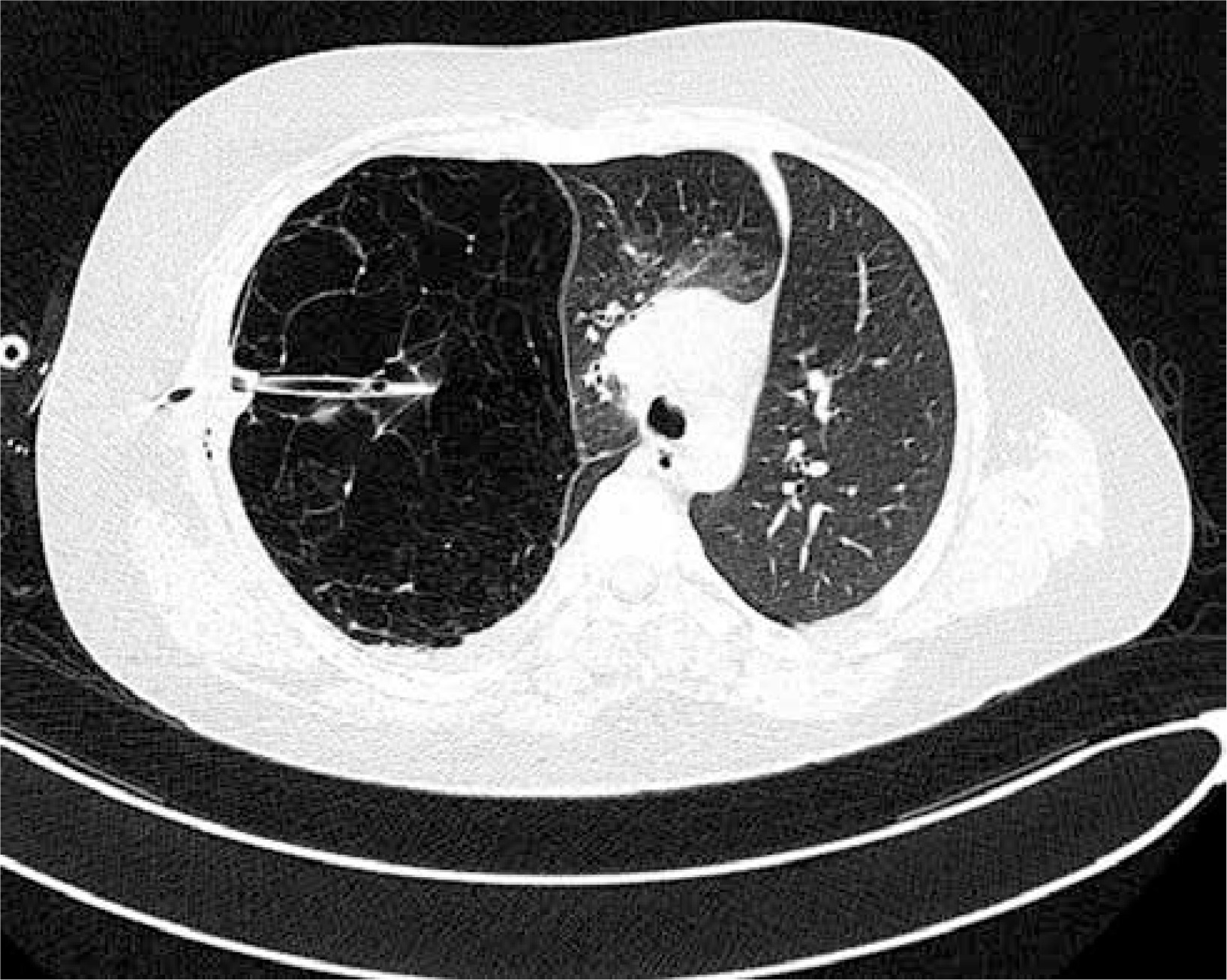
A 13-year old boy, with uneventful past medical history, presented to his GP with the symptoms of breathing difficulty, pain in the chest, shortness of breath, and a dry, nonproductive cough. According to the medical interview, the symptoms had developed over the last 4 days, along with the infection of the respiratory tract, and had progressively worsened. Family history was not significant, and personal pathological history included few upper airway infection episodes, which were treated by the family doctor with symptomatic drugs. Physical examination revealed distended chest with minimal movements on the right side, hushed breathing sounds over the right lung, and oxygen saturation of 88%. The boy was sent for radiological examination. X-ray of the chest showed increased translucency of the right hemithorax with leftward shift of the mediastinum (Figure 1), and was interpreted as right-sided pneumothorax. The boy was immediately admitted to a regional hospital, and underwent chest drainage. The drain was placed in the second intercostal space, right midclavicular line; nevertheless the symptoms did not improve, and the air leak was not observed in the drainage system. The general state of the patient was worsening. The boy was transported to the clinical hospital, where the first drain was removed, and the second drain was inserted in the fifth intercostal space in the right mid axillary line, and suction was applied. This time, constant leakage of air in the drainage system was observed, the general state of the patient rapidly improved, and the symptoms resolved. Closer examination of the first and follow-up X-rays of the chest brought the suspicion of congenital lobar emphysema of the right upper lobe. The CT of the chest showed a bullous change occupying approximately 85% of the right lung and displacing the mediastinum, and revealed that the second drain was inserted into the emphysematous right upper lobe (Figure 2). Bronchoscopy showed narrowing of the right upper lobe bronchus, without pathological secretions. Cystic fibrosis was excluded based on two negative sweat tests.
Figure 1
Initial X-ray of the chest: increased translucency of the right hemithorax with leftward shift of the mediastinum
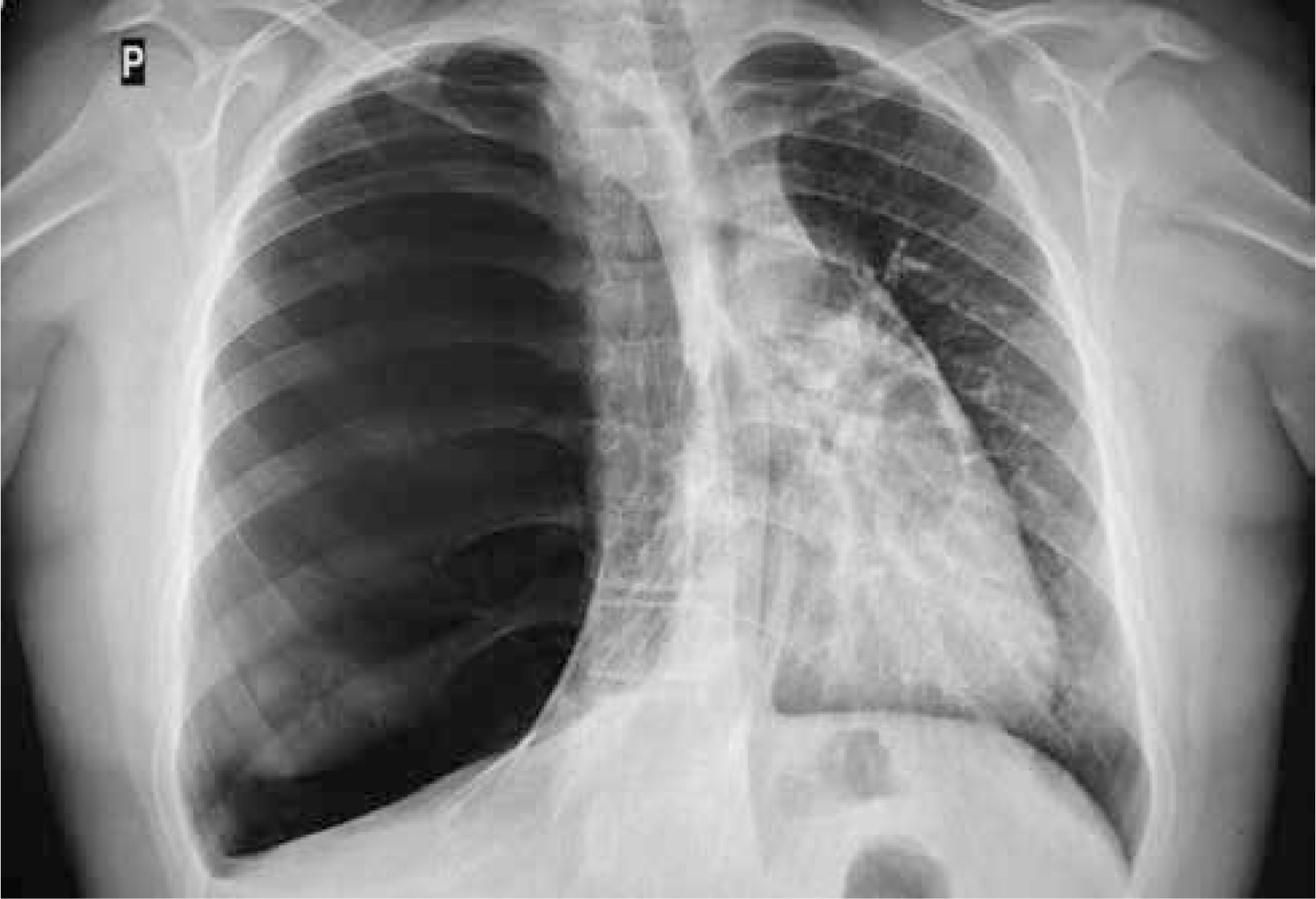
Figure 2
Computed tomography of the chest – collapse of apical and posterior segments of the right upper zone along with hyperinflation of the right middle lobe, causing mediastinal displacement to the left, drain inserted into the emphysematous right upper lobe
The boy was scheduled for thoracoscopic right upper lobectomy. During the procedure the conversion to thoracotomy was made, due to the size of the right upper lobe. We used a TachoSil patch for ensuring air-tight closure of the right upper bronchus. After the procedure we left one chest tube, which was removed 2 days after the surgery. We did not observe any complications; the hospital stay, surgery and recovery were uneventful. The histopathology report was consistent with the diagnosis of CLE. Follow-up X-ray of the chest showed normal expansion of the lungs. The patient was followed up for 1 year. He is in an excellent condition, without complaints during daily and sport activities.
In CLE a defect of the bronchial cartilage leads to air being trapped, because of the valve effect during expiration [1–4]. Defective ventilation leads to progressive respiratory failure [1, 4]. Innate cardiac malformations such as patent ductus arteriosus, atrial septal defect, ventricular septal defect, total anomalous pulmonary venous return, and tetralogy of Fallot can accompany CLE [1]. Incorrect embryo development between 4 and 6 weeks of gestation affect both lungs and heart [2]. CLE in infants and neonates usually manifests with breathlessness, cough, wheezing, and respiratory symptoms [2]. Still, some patients may become symptomatic later in life [5]. Santra et al. reported CLE diagnosed at the age of 15 years, as in our case of an adolescent boy [5]. The diagnosis of CLE requires a high degree of suspicion. CLE may be misdiagnosed as pneumothorax and the treatment with intercostal drainage may be detrimental, as was the case with our patient [5]. The prenatal diagnosis of CLE is uncommon, but USG can show hyperechogenicity in lung segments without abnormal blood flow, shift of mediastinum and polyhydramnios [1, 4]. Congenital cystic adenomatoid malformation and pulmonary sequestration should be considered in the differential diagnosis [1, 2, 4]. Fetal MRI may also be helpful in the diagnostics [1, 4]. Changes at the organ level may result from molecular and biochemical changes [6]. Some authors see a connection between emphysematous changes in the lungs and congenital α1 antitrypsin deficiency [7].
CLE is usually diagnosed with chest radiography. Chest X-ray can show hyperinflation of the affected lobe, and contralateral displacement of the mediastinum [2]. Atelectasis and increased density in the adjacent lobes result from the compression [2]. Bronchoscopy is usually normal, but in our case bronchoscopy showed narrowing of the right upper lobe bronchus, without pathological secretions [1–3]. CT remains the gold standard in the diagnosis of CLE; it can reveal vascular anomalies and mediastinal masses [2]. Angiography can reveal CLE resulting from compression of a vascular structure [2]. The use of bronchoscopy is controversial. In patients with respiratory distress it can worsen the symptoms [1, 4]. Radionuclide imaging can help in planning the treatment [1, 4]. However, it is not recommended by other authors [2].
The treatment of choice is lobectomy [1, 4]. An alternative can be the resection of the affected lobe segment [1, 3, 4]. The most common surgical technique in children is thoracotomy, but video-assisted thoracoscopy is safer and faster [1, 3, 4]. The complication and mortality rates are lower in the pediatric population [1–4]. In bilateral presentation of CLE, usually bilateral lobectomy is unnecessary [1, 4].
During intratracheal mechanical ventilation, positive pressure may exacerbate the symptoms [1, 3, 4]. High-pressure ventilation should not be performed – the airway pressure should not exceed 20–25 cm H2O, and some authors suggest selective intubation [1, 3, 4].
Children with minimal respiratory symptoms and normal bronchoscopy can be treated with observation alone, but follow-up is needed as they may require surgical intervention later on due to sudden respiratory distress, as occurred in our case [1, 4].
Recent studies have demonstrated that in selected patients, a complete single lobar obstruction using an IBV valve can be beneficial in improving the pulmonary function and clinical outcomes [1, 2]. This approach is still controversial, but it can be an option for patients who cannot tolerate surgery.
Diagnosis of congenital lobar emphysema requires a high degree of suspicion. In our patient the misinterpretation of the initial X-ray caused the delay in proper surgical treatment. CT scan is the best diagnostic study. Lobectomy leads to dramatic improvement in the condition of the patient.









