
Introduction
Esophageal squamous cell carcinoma (ESCC) is one of the most aggressive malignancies, with a high mortality rate, particularly in Asian countries. Despite advances in treatment modalities, the prognosis for ESCC remains poor, primarily because of its propensity for immune evasion, lymphatic metastasis, and resistance to apoptosis [1]. For example, a phase III trial showed that in East Asian patients with locally advanced ESCC undergoing concurrent chemoradiotherapy with weekly paclitaxel and carboplatin, followed by two cycles of consolidation chemotherapy with the same drugs, the 3-year progression-free survival and overall survival (OS) rates were 46.4% and 56.5%, respectively [2]. Thus, an abundance of research has been directed toward understanding the complex biochemical and molecular pathways underlying ESCC progression, including those involved in oxidative stress, pro-inflammatory responses, and tumor immune escape mechanisms [3–10].
Oxidative stress and immune-modulatory genes, including high-mobility group box 1 (HMGB1), toll-like receptor 4 (TLR4), interleukin-6 (IL-6), fibroblast growth factor receptors 1 and 2 (FGFR1 and FGFR2), and ROS1 have been implicated in ESCC. HMGB1, a non-histone DNA-binding protein, plays an important role in inflammation and immunomodulation by interacting with the Keap1/Nrf2 pathway to activate NF-κB, thereby promoting indoleamine 2,3-dioxygenase (IDO) expression [3]. This results in an immunosuppressive tumor microenvironment conducive to ESCC development. HMGB1 also interacts with TLR4, which is an important immune receptor whose upregulation has been associated with enhanced lymphatic metastasis in ESCC [11].
The interactions of ROS1 with other genes highlight its role in modulating oxidative stress and influencing the immune response in the tumor microenvironment [12]. ROS1, as a receptor tyrosine kinase, contributes to cell proliferation and apoptosis resistance through redox imbalance and activation of pro-survival signaling pathways. Recent studies have shown that it can further potentiate tumorigenesis by upregulating pathways associated with oxidative stress and recruiting immunosuppressive cells, thus contributing to immune evasion [13]. In addition, FGFR1 and FGFR2 are involved in various signaling pathways, including the PI3K-Akt and SHP2 pathways, which enhance cell survival, and are directly implicated in worse survival outcomes in ESCC patients [10, 14–17]. Fibroblast growth factor receptor 1 has also been shown to correlate with significantly decreased overall survival, making it a potential prognostic marker [9, 10, 15, 18].
Despite numerous studies into each of these genes, studies examining their combined effects in ESCC are limited. Therefore, in this study, we examined the interactions among these key oxidative stress-related and immune-modulatory genes (HMGB1, TLR4, IL6, FGFR1, FGFR2, and ROS1) to better understand their roles in ESCC progression and potential as therapeutic targets.
Materials and methods
Ethics considerations
The Kuang Tien General Hospital General Hospital Institutional Review Board reviewed and approved the request that the dataset be de-identified and that the study qualifies under the exempt categories. Approval was granted under approval number KTGH-11338, and the requirement for patient informed consent was waived.
Datasets and patient selection
Supplementary Figure S1 provides an overview of the alteration frequency and genomic data types available from five studies involving ESCC patients and includes data on mutation types, copy number aberrations (CNA), and structural variants, organized by study. The eligible patients and genomic data were compiled from five key datasets: TCGA, Nature [19]; TCGA, Firehose Legacy, Broad Institute [20]; MSK, J Natl Cancer Inst 2023 [21]; ICGC, Nature 2014 [22]; and UCLA, Nat Genet 2014 [23]. All five studies include mutation data, CNA data are available in four studies, and structural variant data are provided in three studies. These comprehensive datasets represent a valuable resource for analyzing genetic alterations in ESCC, with diverse genomic information available from multiple high-impact studies. Data from 1,873 patients (1,874 samples) diagnosed with various esophagogastric cancers were reviewed and 437 pa- tients with pure ESCC were selected for further analysis. In addition, the gene expression omnibus ESCC dataset (GSE53624) was used to examine the correlation between gene expression levels and survival, with a focus on six genes [24]. GSE45670 was used to analyze the fold change of the six genes’ expression levels compared with the normal esophageal epithelium as a predictive biomarker for pathological complete remission (pCR) [25].
Merging five datasets
Through the cBioPortal platform [26–28], the five datasets were harmonized to extract genomic alterations in six genes of interest: HMGB1, ROS1, IL6, FGFR1, FGFR2, and TLR4. The merging process ensured consistent categorization of mutation types (e.g., missense mutations, truncating mutations), copy number alterations (amplifications and deletions), and structural variants in the datasets, which were visualized using an OncoPrint format.
Data normalization
Normalization is crucial for any continuous or count-based data that can vary due to technical factors, such as gene expression (RNA-seq, microarray), DNA methylation, and copy-number data, because these measurements rely on procedures such as scaling, log transformation, and batch correction to ensure accurate, comparable results. In contrast, mutation data (e.g., variant call format or mutation annotation format files) generally do not require “expression-style” normalization, since they consist of discrete variant calls rather than continuous signals, though they may still undergo quality control and representation “normalization” (e.g., left-aligning indels).
Normalization pipelines differ widely by technology (RNA-seq vs. microarray vs. methylation, etc.). cBioPortal is designed primarily for visualization and integrative analyses, and therefore it avoids adding a “black box” step by expecting data providers to perform their own normalization. According to the cBioPortal user documentation and data-loading guidelines, once already-processed and normalized data are submitted, cBioPortal optionally computes per-gene Z-scores (mean of zero, standard deviation of one) across samples for visualization and outlier detection, relying on the data to be analysis-ready (for example, transcripts per million, reads per kilobase of transcript per million mapped reads, or variance stabilizing transformation in differential expression sequencing 2 for RNA-seq, or robust multi-array average/quantile normalization for microarrays). cBioPortal also requires the data to be in a single scale (e.g., log2) and consistent across all samples, and if a “normal” subset of samples is specified, it can calculate Z-scores relative to those normal samples. Finally, users must provide these normalized values in specific file formats, thereby ensuring that the data is uniform, properly scaled, and suitable for cBioPortal’s visualization and analysis tools.
Sorting patients
Patients were categorized based on the presence of any genetic alteration in the six genes of interest. The following two groups were established: the altered group – patients with at least one alteration in any of the six genes (n = 66; 15%); and the unaltered group – patients without alterations in these genes (n = 371; 85%). Demographic, clinical, pathological, and molecular data were collected and compared between the groups to evaluate associations with clinical outcomes.
Query in the cBioPortal and two other independent cohorts: ESCCdb (Sichuan) and GSE45670
Data mining was performed using the cBioPortal platform (accessed November 2024) and the ESCCdb from Sichuan and GSE45670 [25], which enabled the retrieval of specific genomic alterations and expression data for analysis.
Statistical considerations and power estimation
The Wilcoxon test was used to compare non-parametric numerical data between the altered and unaltered groups, including age at diagnosis, mutation count, and tumor mutation burden. The χ2 test was applied to categorical data, including sex, tumor location, and cancer stage, to assess the distribution of clinical characteristics between the groups. Kaplan-Meier survival curves were generated to compare disease-free survival (DFS) and OS between the altered and unaltered groups. The log-rank test was used to assess the statistical significance of differences between survival curves. Hazard ratios with 95% confidence interval (CI) were calculated to quantify the relative risk of disease recurrence (for DFS) and mortality (for OS) in the altered group compared with the unaltered group. The α level for statistical significance was set at 0.05. These methods provided a structured approach to assess the impact of specific gene alterations on the clinical outcomes of ESCC patients.
With an α level of 0.05 and a true hazard ratio of 2.0, the estimated power to detect differences in OS between the altered and unaltered groups is 99.82%. This indicates that the study is highly likely to detect significant survival differences between the groups, even with unequal group sizes. STATA 18.0 was used for power estimation. (StataCorp. 2023. Stata Statistical Software: Release 18. College Station, TX: StataCorp LLC.)
Results
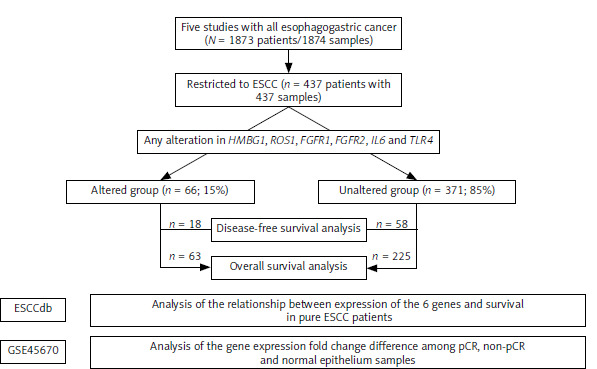
Figure 1 illustrates the study flowchart for selecting patients with ESCC from five merged datasets. The study began with 1,873 patients (1,874 samples) diagnosed with various types of esophagogastric cancer. Based on this cohort, 437 patients with ESCC (437 samples) were selected. They were categorized based on genetic alterations in at least one of the six genes of interest. The altered group consisted of 66 patients (15%), while the unaltered group included 371 patients (85%). The flowchart provides an overview of patient selection and the categorization used for survival analysis based on the presence or absence of specific gene alterations in ESCC.
Fig. 1
Flowchart depicting the selection of patients with esophageal squamous cell carcinoma merged from five datasets; and analyses using two independent transcriptome datasets
ESCC – esophageal squamous cell carcinoma, pCR – pathological complete remission
OncoPrint was used to visually summarize the genomic alterations among the six genes, with each block representing an individual patient (Suppl. Fig. S2). The alterations included: missense mutations (colored sections within each block), which indicate specific amino acid changes that may affect protein function; truncating mutations – typically more disruptive as they lead to incomplete proteins; amplifications (red) – exhibit increased gene copy numbers, which may result in overexpression of these genes and contribute to tumor progression; deep deletions (blue) – show loss of gene copies, potentially leading to loss of function for tumor suppressor roles. The alterations varied, with HMGB1 showing alterations in approximately 14% of patients. ROS1 exhibited alterations in 6% of the cases, whereas TLR4 showed alterations in 2% of the cases. Fibroblast growth factor receptors 1 and FGFR2 had higher alteration rates of 8% and 7%, respectively, which often included amplifications. Interleukin-6 alterations were observed in 2% of the patients. OncoPrint (Suppl. Fig. S2), with data aggregated from five different ESCC studies, provides a comprehensive view of how frequently these genes undergo specific types of genetic alterations.
Supplementary Figure S3 displays the pairwise analysis of mutual exclusivity or co-occurrence among the six genes of interest in ESCC. Co-occurrence was observed using the following gene pairs: HMGB1 and ROS1, with a significant p-value (0.009) and a high log2 odds ratio (> 3). ROS1 and FGFR1 had a log2 odds ratio of 1.879, although the association did not reach statistical significance (p = 0.080). FGFR1 and FGFR2 showed weak evidence for co-occurrence, with a log2 odds ratio of 2.258 (p = 0.105). Mutual exclusivity was suggested for other gene pairs, as indicated by a log2 odds ratio < –3. The analysis highlights certain interactions in which genes may be more likely to occur together within patients with ESCC, potentially pointing to shared pathways or cooperative roles in tumorigenesis.
Table 1 comprehensively compares the demographic and clinical data between the altered and unaltered groups. Although the altered group was slightly younger, there were no major demographic or clinical characteristics showing a strong association with the genetic alteration status. The median age was slightly younger in the altered group (57 years) compared with the unaltered group (60 years), with a significant difference (p = 0.0239). The majority were male in both groups, with 86.36% in the altered group and 77.09% in the unaltered group, showing no significant difference (p = 0.953). Both groups had similar median body weights, with no significant difference (p = 0.650). The altered group smoked a median of 25 pack-years, slightly less than the 30 pack-years in the unaltered group (p = 0.0912). Alcohol consumption was not significantly different between the two groups (p = 0.346). The performance status, as measured by the Karnofsky score, was similar in both groups, with no significant difference (p = 0.903). The distribution of tumor location (proximal, middle, distal) showed no significant difference (p = 0.400). The overall stage distribution between the groups showed no significant differences (p = 0.180). There was no significant difference between groups in the use of adjuvant postoperative radiotherapy (p = 0.513), nor was a significant difference observed in the use of adjuvant systemic therapy (p = 0.854).
Table 1
Comparison of demographic and clinical data between the altered and unaltered groups
Table 2 compares the pathological and molecular data between the altered and unaltered groups. There were key molecular differences between the groups, particularly in genome alteration, mutation count, and tumor mutation burden, which were significantly higher in the altered group. The altered group had a higher median altered genome fraction (0.43) compared with the unaltered group (0.37), showing a significant difference (p = 0.0474). The mutation rate was slightly higher in the altered group (6.35%) compared with the unaltered group (5.68%), although the difference was not statistically significant (p = 0.105). The altered group had a significantly higher mutation count (median 158.5) compared with the unaltered group (median 56), with a highly significant difference (p < 1E-10). The altered group also had a higher non-synonymous tumor mutation burden (median 5.33) compared with the unaltered group (median 3.37), which showed a significant difference (p = 5.22E-8).
Table 2
Comparison of pathological and molecular data between the altered and unaltered groups
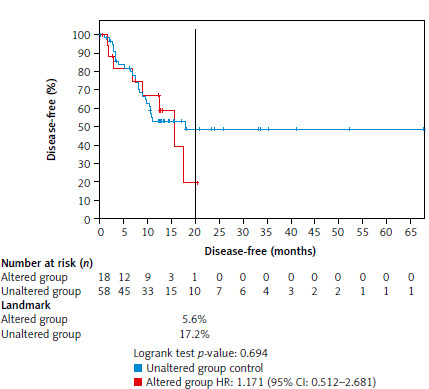
Figure 2 shows the DFS comparison between the unaltered (reference) and altered groups in the ESCC patients, with the follow-up extending beyond 65 months. At the 20-month mark, 17.2% of the patients in the unaltered group remained disease-free compared with only 5.6% in the altered group, indicating a lower DFS rate in the altered group. The altered group had a nonsignificantly higher risk of relapse compared with the unaltered group, with a hazard ratio of 1.17 (95% CI: 0.512–2.681). The log-rank test yielded a p-value of 0.694, indicating that the observed difference in DFS between the two groups was not statistically significant. These data suggest a tendency for poorer outcomes in patients with gene alterations, although further studies with a larger sample size would be necessary to confirm this.
Fig. 2
Comparison of disease-free survival between the unaltered group (reference group) and the altered group with a follow-up of more than 65 months
At 20 months, 17.2% in the unaltered group were still disease-free compared with only 5.6% in the altered group. The altered group showed an nonsignificant increased risk of relapse with a hazard ratio of 1.17 (log-rank, p-value = 0.694).
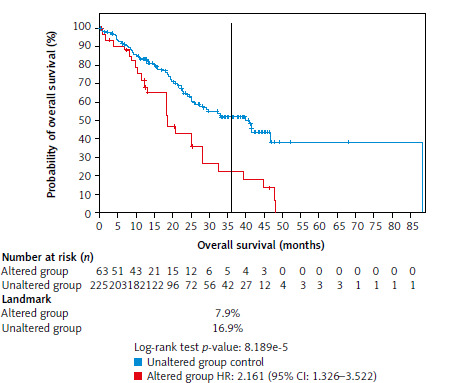
The overall survival between the unaltered (reference) and altered groups in patients with ESCC, with follow-up extending beyond 85 months, is presented in Figure 3. At the 36-month mark, 16.9% of patients in the unaltered group were alive, compared with only 7.9% in the altered group. The altered group exhibited a significantly increased mortality risk with a hazard ratio of 2.16 (95% CI: 1.326–3.522), suggesting that patients with alterations in the six genes of interest had a greater than twofold higher mortality risk compared with the unaltered group. The log-rank test yielded a highly significant p-value of 0.000082, indicating a statistically significant difference in OS between the altered and unaltered groups.
Fig. 3
Comparison of overall survival between the unaltered group (reference group) and the altered group with a follow-up of over 85 months
At 36 months, 16.9% in the unaltered group were still alive, compared with only 7.9% of the altered group. The altered group showed a 2.2-fold increased risk of death, with a hazard ratio of 2.16 (log-rank, p-value = 0.000082).
Supplementary Figure S4 shows the relationship between gene expression and survival in the gene expression omnibus ESCC dataset (GSE53624). Low expression of HMGB1 (defined as expression levels at or below the first quantile) was significantly associated with better OS compared with higher expression levels (above the third quantile) in a follow-up period of 70 months. This association was statistically significant, with a log-rank p-value of 0.008. This suggests that lower HMGB1 expression may contribute to improved survival outcomes in ESCC, indicating its potential role as a prognostic biomarker. Subsequently, we queried these six genes in a different cohort of ESCC (GSE45670) who had pretreatment gene expression analysis and were treated with preoperative chemoradiotherapy before radical resection with or without a pCR. HMGB1 expression in the pCR group was significantly higher than in the normal (esophageal epithelium) group, p = 0.016 (Dunn test) (Suppl. Table S1). There were no significant differences between normal vs. non-pCR or non-pCR vs. pCR.
Discussion
The key findings of this merged dataset study indicate that approximately 15% of patients with ESCC harbor alterations or co-occurring alterations in the six genes of interest: HMGB1, ROS1, FGFR1, FGFR2, IL6, and TLR4. These genomic alterations are associated with significantly poorer long-term survival outcomes. Although prior studies have explored the roles of these genes individually in ESCC, this study is the first to present patient-level evidence of their collective impact on patient prognosis. Over an extended follow-up period exceeding 85 months, the altered group demonstrated a statistically significant twofold increase in mortality risk. Although the difference in DFS did not reach statistical significance because of limited relapse data, worse DFS outcomes were observed in the altered group. These findings reinforce and build upon previous individual studies examining the role of each gene in ESCC, providing a more comprehensive understanding of their potential combined effects on patient survival.
Based on the current study, the effect of the six genes on survival was not only the result of individual gene expression. Other types of genomic alterations, such as amplifications, deep deletions, and mutations, may also play significant roles, individually or in combination. These alterations can affect downstream pathways, such as NF-κB and other immune-evasive pathways, which contribute to tumor progression and resistance to the immune response. The mechanistic explanation of how the altered genes are involved in the oxidative stress-related pathway and the PD-1/PD-L1 pathway is relatively complex. For example, the HMGB1 cascade may interact with the Keap1/Nrf2-ARE pathway [29, 30], and the common pathway of the Keap1/Nrf2/NF-κB/IL-6 axis promotes tumor immune evasion [31, 32]. The HMGB1/TLR4/MYD88/NF-κB pathway was shown to induce autophagy, promote proliferation, inhibit apoptosis, and enhance radioresistance in ESCC [33, 34]. The poor prognosis associated with alterations in HMGB1, ROS1, FGFR1, FGFR2, IL-6, and TLR4 in ESCC may be attributed to the involvement of these genes in multiple oncogenic pathways that promote tumor aggressiveness and immune evasion (Suppl. Table S2).
Recently, due to the efficacy of adding an immune checkpoint inhibitor in managing ESCC [35], there has been growing interest in identifying an easily assessable immune biomarker, specifically the pan-immune-inflammatory value (PIV), to correlate with patient treatment outcomes. PIV is calculated using the formula: (neutrophil count × platelet count × monocyte count)/lymphocyte count [36]. A Japanese study found that a low PIV, defined as a value below 164.6, was associated with higher tumor-infiltrating lymphocytes and CD8+ cell counts, as determined by immunohistochemical analysis, as well as improved OS [37]. However, our analysis indicates that lymphocyte infiltration within the tumor, as assessed by pathologists through microscopic evaluation, does not significantly differ between the altered and unaltered groups. Future prospective studies incorporating PIV may provide more robust evidence regarding its clinical utility.
HMGB1, a key mediator of inflammation, significantly contributes to tumor progression by activating the receptor for advanced glycation end products and toll-like receptors (TLRs), particularly TLR4 [11]. This interaction stimulates the NF-κB signaling pathway, which increases the production of pro-inflammatory cytokines and promotes a tumor-permissive microenvironment. In addition, HMGB1 enhances the expression of indoleamine IDO, leading to immunosuppression and facilitating immune escape by modulating T-cell function in the tumor microenvironment [3].
ROS1, another important player, acts as a receptor tyrosine kinase involved in oxidative stress regulation and cell proliferation. Its activation enhances redox imbalance, which fosters survival signaling and apoptosis resistance. Studies indicate that ROS1, along with other receptor tyrosine kinases, such as FGFR1 and FGFR2, can drive tumor growth by activating the PI3K-Akt and Ras-MAPK pathways, resulting in enhanced tumor cell proliferation and migration [15]. The effect of FGFR1 and FGFR2 is further underscored by their role in autophagy and Nrf2 activation, which enhance resistance to cellular stress and support the invasive potential of tumor cells [17]. Increased FGFR1 expression is associated with reduced survival; thus, it is a potent prognostic marker for aggressive disease [38].
Interleukin-6 and TLR4 add another layer of complexity by reinforcing inflammatory signaling within the tumor. Interleukin-6 promotes cancer cell survival, particularly through the JAK/STAT and NF-κB pathways, which are essential for maintaining chronic inflammation and immune resistance. In ESCC, elevated IL-6 expression correlates with poor survival, as it recruits myeloid-derived suppressor cells and promotes immune escape mechanisms [39]. On the other hand, TLR4 is a main receptor of innate immunity and acts as a sentinel receptor that triggers pro-inflammatory responses, leading to lymphatic metastasis and further contributing to an immune-suppressive environment [33, 40].
Fibroblast growth factor receptors FGFR1 and FGFR2 play distinct yet complementary roles in ESCC. FGFR1 amplification, found in up to 21% of ESCC cases, promotes tumor growth, invasion, and poor prognosis by activating the MEK-ERK and PI3K-AKT pathways [41, 42]. Co-expression of FGFR1 with its ligands further enhances tumor proliferation via an autocrine loop [43]. Conversely, FGFR2 exhibits a dual role in ESCC. While FGFR2 amplification is less frequent (~ 4%), it contributes to resistance against EGFR-targeted therapy [44, 45]. Fibroblast growth factor receptors 2 also maintains cancer cell differentiation through AKT signaling, preventing epithelial-mesenchymal transition [14]. However, excessive FGFR2 signaling promotes tumor progression, as miR-671-5p downregulation leads to FGFR2 upregulation and enhanced proliferation [46]. Together, FGFR1 drives aggressive ESCC phenotypes, while FGFR2’s context-dependent roles highlight its potential as both a therapeutic target and differentiation regulator. Aberrations in FGFR1-3 may be actionable through a histology-agnostic approach using FGFR-targeted therapies. While FGFR inhibitors have not yet been approved specifically for ESCC, preclinical studies suggest that FGFR2-amplified or overexpressing ESCC could respond to these agents [44, 45]. Clinical trials evaluating FGFR inhibitors in ESCC and other FGFR-driven cancers are ongoing.
These combined molecular events orchestrate a network of oxidative stress and immune evasion pathways that likely contribute to the observed poor prognosis in patients with alterations in these six genes. By promoting cell proliferation, reducing apoptosis, and supporting immune evasion, these pathways create a robust environment for tumor progression, which underscores the clinical significance of targeting these pathways for therapeutic intervention.
The strength of this study lies in the integration of multiple genomic datasets, which combine patient data and ESCC specimens across various sources. This approach enhances the study’s statistical power, providing a robust capability to detect true differences in survival outcomes with high significance. Despite the strengths of this study, there are notable limitations. First, the study relied on in silico analyses and public databases, which may introduce inconsistencies in data collection and reporting across sources. Variability in sequencing techniques, sample processing, and annotation methods between datasets may affect the uniformity of genomic alterations observed. In addition, while pooling datasets increases sample size, it may also obscure unique cohort-specific characteristics, potentially limiting the generalizability of the findings. The observational nature of the study further restricts causal inference. Despite the high power for detecting mortality risk, the smaller sample size in the altered group may have limited the ability to achieve significance in DFS analysis. A notable limitation is that survival analyses were conducted using cBioPortal, which only supports univariate Kaplan-Meier analyses and does not allow multivariate Cox regression to adjust for potential confounders such as cancer stage. Finally, while this study provides insight into associations between genomic alterations and survival, functional validation through laboratory-based studies is necessary to confirm the suggested biological mechanisms. These limitations should be addressed in future studies to strengthen the clinical relevance of the findings.
Conclusions
This study aimed to explore the impact of genomic alterations for six selected genes (HMGB1, ROS1, FGFR1, FGFR2, IL6, and TLR4) on the survival outcomes of patients with esophageal squamous cell carcinoma. Using in silico analyses of multiple genomic datasets, we observed that approximately 15% of patients harbored alterations in these genes. These alterations were associated with significantly poorer OS, with a hazard ratio of 2.16 over an extended follow-up period. Although previous studies have individually examined the role of these genes in ESCC, our study provides novel insight into their collective impact, highlighting an increased mutation burden and altered tumor microenvironment as contributing factors to immune evasion and disease progression.
Our findings underscore the relevance of these genes in modulating pathways that support tumor survival and immune resistance, such as the NF-κB and PD-1/PD-L1 pathways. Nonetheless, this is an observational study, and its reliance on public datasets is a limitation, including potential inconsistencies in data collection and cross-cohort variability. Future studies should focus on experimental validation and functional studies to elucidate the underlying mechanism of these genomic alterations and their therapeutic implications.
Overall, our work suggests that the combined genomic alterations in HMGB1, ROS1, FGFR1, FGFR2, IL6, and TLR4 may serve as prognostic markers and potential targets for tailored therapeutic strategies in ESCC, which warrants further studies to enhance the clinical outcomes for this aggressive cancer.






