
Introduction
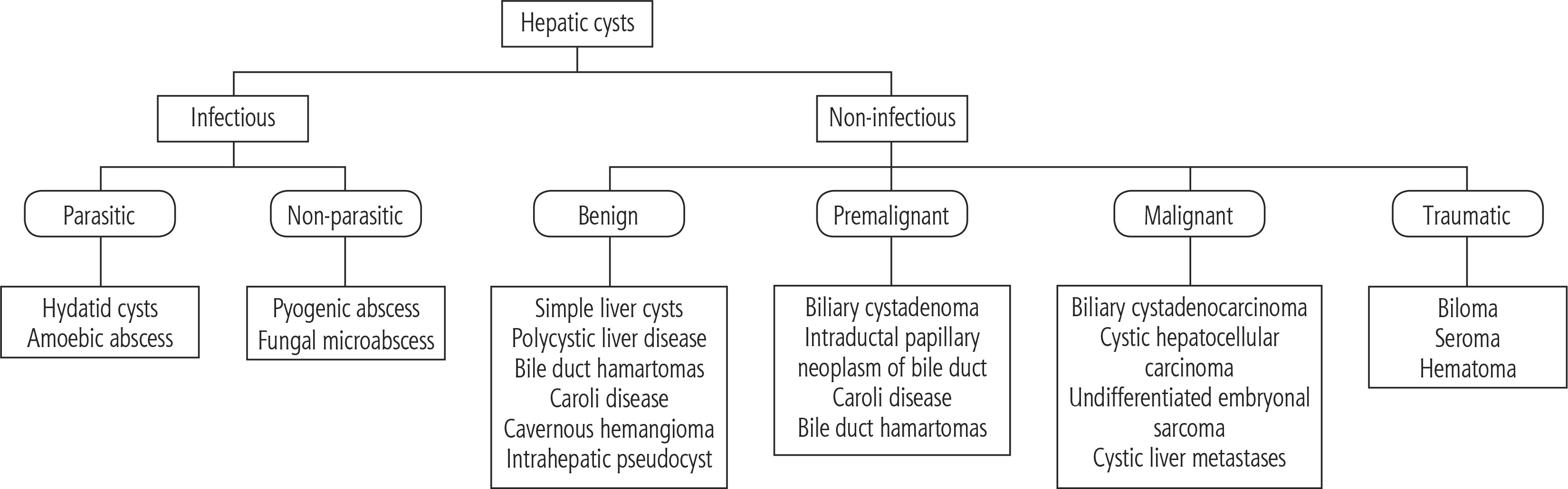
Cystic hepatic lesions are a group of heterogeneous lesions encountered in daily clinical practice. These lesions vary concerning pathogenesis, clinical presentation, and radiological findings. The diagnosis may range from benign cystic lesions to malignant and potentially lethal conditions [1]. Cystic lesions of the liver can be broadly classified as infectious and noninfectious lesions. The main parasitic lesion of the liver is the hydatid cyst, and the non-parasitic lesions are again divided into benign, pre-malignant and malignant and traumatic lesions (Fig. 1) [2]. The most commonly encountered lesion is a simple hepatic cyst, but in addition to its morphology and number, determination of a solid component in the lesion through radiological imaging helps in the diagnostic approach. Except for simple cyst and polycystic liver that can be confidently diagnosed by ultrasound, the use of contrast-enhanced computed tomography (CT) or magnetic resonance imaging (MRI) is essential for definitive or reasonable differential diagnosis [1]. Though modalities such as CT, MRI, and ultrasonography (USG) provide ambiguous results, the utilization of other diagnostic procedures such as serodiagnostic tests and microbubble contrast-enhanced ultrasound (CEUS) are invaluable in differentiating complex cysts, echinococcosis, and cystadenoma/cystadenocarcinoma [2]. These diagnostic techniques also reduce the need for an invasive procedure. Genetic testing for patients with adult polycystic kidney disease (ADPKD) and polycystic liver disease is also available but is not performed on a regular basis, as it does not interfere with management. In conclusion, an amalgamation of recent advances helps in the formulation of a diagnostic algorithm for clinical decision making.
Epidemiology
Before the use of diagnostic imaging, hepatic cysts (HCs) were discovered surgically. A study conducted between 1954 and 1971 found the incidence of HCs to be 17 in 10,000 cases [3]. The prevalence of hepatic cyst has been reported to be as high as 15-18% in the United States. Simple cysts are one of the most common, found in 2.5-18% of the population. Congenital cysts are found more commonly in females of age 40-70 years, whereas acquired cysts (including hydatid, traumatic and inflammatory cysts) are more common in males of age 30-50 years [4]. No more than 10-15% of patients have symptoms that bring the cyst to clinical attention. Mild elevation of liver enzymes such as alkaline phosphatase and γ-glutamyl transferase is observed. Evidence suggests that elevated levels of cancer antigen 19-9 (CA 19-9) in the cystic fluid does not correlate with malignant lesions.
A study by Wang et al. evaluated 21 cases of mucinous cysts, of which 57% were CA 19-9-positive. Interestingly, there was not much significant difference found in CA 19-9 content between the benign biliary cystadenomas (BCAs) and the malignant biliary cystadenocarcinomas (BCACs) [5]. A larger sample size is necessary to characterize the relationship between CA 19-9 and malignancy in HCs. In the current review, we discuss the pathophysiology and clinical management of cystic lesions of the liver.
Simple cyst
The benign cyst is the most common pathology of the liver, with a prevalence of 2.5-5%, and a slight female predominance (female-male ratio, 1.5 : 1) [6]. The prevalence of solitary cyst in the general population ranges from 2.5% to 18% [7]. They occur as simple fluid-filled structures, with a diameter of < 1 cm up to 30 cm and scattered on both sides of the liver. The simple cyst has thin walls, lined by cuboidal epithelium, and up to two septa [3]. Simple cysts include congenital cysts, Caroli disease, biliary hamartomas and polycystic liver disease (PCLD).
Pathogenesis
The majority of simple cysts are congenital and form from biliary ducts that do not connect to the biliary system. Biliary hamartomas are derived from the embryonic bile ducts. In the case of Caroli disease, which is an autosomal recessive disorder, cavernous ectasia of the bile ducts is observed. However, cysts in Caroli disease have about a 7% chance of developing into cholangiocarcinoma [8]. Two mechanisms have been proposed for cyst formation in PCLD. The mechanism of cyst formation in PCLD is not entirely understood. The first mechanism is thought to be due to retained abnormal bile ductules, which then become detached from the biliary tree and progressively dilate, forming cysts. An alternative mechanism is a defect in biliary cilia, leading to hyperproliferation of cholangiocytes and generation of cysts [9].
Clinical features
A small fraction of patients present with abdominal pain, early satiety, nausea, and vomiting, which arise as a result of a mass effect. Physical examination may reveal a palpable mass in the abdomen or hepatomegaly. Complications such as hemorrhage, rupture and biliary obstruction are uncommon but are more likely in larger cysts. Intracystic hemorrhage is a rare complication of simple cysts and usually presents with severe abdominal pain [10], although asymptomatic presentations are also observed.
Investigation
Laboratory findings are normal, except a minority who demonstrate elevated levels of γ-glutamyl-transferase (GGT). Carcinoembryonic antigen (CEA) and CA 19-9 may be elevated. CA 19-9 is expressed in the inner epithelial lining of a simple cyst and leads to increased cyst fluid and serum CA 19-9 levels [11].
Diagnostic features
Most simple liver cysts are found incidentally during imaging for other reasons. The following USG criteria are suggestive of a simple cyst: anechoic (i.e., fluid-filled cavity), no septations, smooth borders, sharp, strong posterior wall echoes (indicating a well-defined fluid/tissue interface), oval or spherical shaped and relative accentuation of echoes beyond the cyst compared to echoes at similar depth transmitted through the normal adjacent hepatic tissue [12]. USG has a sensitivity and specificity of approximately 90% for diagnosing simple cysts, and recent advances in CT and MRI technology might result in even higher sensitivity rates [8]. However, due to the radiation load associated with CT and high cost involving CT and MRI, USG remains the most accurate, cost-effective imaging and non-invasive modality for diagnosing simple cysts. The recent development of microbubble contrast-enhanced ultrasound (CEUS) enables us to visualize vascular flow within septa or solid components of cysts, which is absent in simple cysts with intracystic hemorrhage [13]. Therefore, CEUS can accurately characterize these cysts when USG, CT, and MRI show ambiguous features.
Treatment
Treatment of a symptomatic cyst involves either percutaneous aspiration or surgical intervention. The addition of a sclerosant such as tetracycline, ethanol, or ethanolamine during aspiration improves outcomes. There are concerns of high failure rate and rapid recurrence with percutaneous aspiration [4]. One review that compared aspiration sclerotherapy with fenestration reported similar failure rates (4 out of 91 vs. 5 out of 85 cases) but showed that the incidence of adverse incidents is slightly lower after aspiration sclerotherapy [14].
Surgical techniques involve cyst fenestration, subtotal cyst excision, liver resection, and even liver transplantation. Studies comparing the outcomes of these conventional surgical techniques are hampered due to the multiple treatment algorithm, small patient numbers and relatively short follow-up periods [15]. The laparoscopic approach is the current standard of care, but studies documenting long-term follow-up after laparoscopic treatment of simple liver cysts have shown recurrence rates varying from 4% to 41% [16]. A meta-analysis of 31 studies performed by Zhang et al. found no difference in recurrence rates between the open fenestration group and the laparoscopy group, regardless of whether it was a single or multiple hepatic cysts [17].
Echinococcal cyst
Echinococcal cyst (EC) or hydatid disease is a parasitic infection caused by Echinococcus granulosus. Definitive hosts for E. granulosus are dogs, with sheep being the major intermediate host. However, cattle, horses, pigs, goats, and camels are also potential intermediate hosts. People are only incidentally infected when they ingest tapeworm eggs [18].
Pathogenesis
The ingested parasitic eggs hatch in the small intestine. The resulting larvae infiltrate the blood and lymphatic circulation system. Then, they travel through the portal vein into the liver, lungs, and other tissues, and the larvae develop into hydatid cysts. The liver is the most common site for cystic lesions (52-77%) seen in hydatid disease, followed by lung (10-40%), brain, and other viscera [19]. The cyst is slow growing and exists subclinically for several years.
Clinical features
Diagnosis is made in part by clinical history with attention paid to patient’s residence, place of origin and occupation to identify high-risk patients. The most common symptom is pain in the right upper quadrant, or the epigastrium, whereas a palpable mass and hepatomegaly are the most common signs. Non-specific symptoms such as nausea, fever or dyspepsia may be present. Complicated hepatic hydatid disease may present with signs and symptoms of fever, jaundice, or anaphylactic symptoms [18]. Rupture of a hydatid cyst may cause fever, pruritus, eosinophilia, or fatal anaphylaxis. Acute cholangitis is seen in the case of rupture into the biliary tract and bilioptysis in the case of bilio-bronchial fistula [20].
Diagnosis
Diagnosis of echinococcal cyst involves both serological evaluation and imaging modalities. Serologic tests such as latex agglutination, hemagglutination and enzyme-linked immunosorbent assay (ELISA) are associated with a high incidence of false-negative and false-positive results [21]. Specific IgE antibodies are demonstrated with ELISA. The radioallergosorbent test is positive in the presence of active hydatid disease. Confirmatory tests such as arc-5 immuno-electrophoresis and immunoblotting use parasite-specific antigens. The positivity rate with the arc-5 immunoelectrophoresis is as high as 91.1% [19]. USG and CT are the first choices of imaging in the diagnosis of EC, with the specificity of USG in the range of 90%. CT is helpful for confirming the diagnosis and can reveal calcified cystic walls, daughter cysts, and exogenous cysts, as well as evaluating the cyst volume and density. Four different radiographic appearances have been described on imaging: simple cyst with no internal architecture, cyst with daughter cysts and a matrix, calcified cyst, and complicated cyst [19]. CT and MRI have very high specificity and sensitivity in the detection and differential diagnosis of hepatic cysts and extracapsular (satellite) cysts. Accurate diagnosis of ECs is essential because the mortality of these lesions is slightly higher than for simple cysts, estimated at 2-5% [22].
Treatment
The goal of treatment in the case of hepatic hydatid disease involves elimination of the parasite and prevention of recurrence of disease with minimum morbidity and mortality. Three therapeutic options are currently in use; these involve systemic chemotherapy with mebendazole/albendazole, surgery, and the treatment known as “puncture, aspiration, injection, re-aspiration” (PAIR). Surgical intervention consists of open conservative, radical, and laparoscopic approaches [18]. Conservative techniques involve drainage, marsupialization, deroofing and open or closed total cystectomy with or without omentoplasty. Radical procedures used are total pericystectomy, partial hepatectomy, and lobectomy. Whichever techniques are used, a benzimidazole agent is administered before any surgery in an attempt to sterilize the cyst and reduce the risk of anaphylaxis [18]. PAIR involves puncture, aspiration of the cyst, injection of hypertonic saline and/or absolute alcohol, and re-aspiration. PAIR treatment satisfies all the treatment goals of surgery in hydatid disease but substitutes germinal membrane sclerosis and separation using scolicidal agents for surgical removal. PAIR drainage is best performed under continuous ultra-sonographic or CT guidance with benzimidazole coverage. A study by Chen et al. showed that clinical and parasitological cure in patients undergoing laparoscopic intervention was 98.7% and in patients receiving PAIR plus chemotherapy was 97.5% [23].
Biliary cystadenoma
Biliary cystadenoma (BCA) is the most common premalignant lesion in the liver. It is a rare, slow-growing neoplasm arising from the bile ducts. The pathogenesis is that these lesions arise from ectopic remnants of embryonic bile ducts or aberrant ducts. The presence of endocrine cells in BCAs and cystadenocarcinomas is consistent with an origin from peribiliary glands.
Epidemiology
The incidence of BCA is 1-5/100,000 people. Even though previously published literature quoted the incidence of BCAs to be around 5% of cystic hepatic tumors, still the actual incidence is controversial. Female preponderance is noted (female : male ratio, 9 : 1), with the mean age of presentation being 45 years [24]. BCA usually arise from the intrahepatic bile duct although BCAs of the extrahepatic biliary tract and gallbladder have been reported in 10-20% of cases. The reported rate of malignant transformation to cystadenocarcinoma can be as high as 30% [25].
Clinical features
Patients are usually asymptomatic or present with non-specific symptoms. Diagnosis of BCA remains challenging due to the presence of a wide variety of cystic lesions in the liver including simple cysts, parasitic cysts, degenerated metastatic tumors, mucin-producing metastatic tumors, and congenital cystic dilation. A lack of established criteria for the preoperative diagnosis of unilocular BCA further complicates matters [26]. Macroscopically, BCA demonstrates a multi-locular cyst filled with mucinous fluid and divided by irregular thick walls; they may vary in size from 1.5 to 35 cm. Edmondson initially defined BCA as a multilocular cyst with ovarian stroma, but BCAs without ovarian stroma were subsequently reported. This, in turn, led the WHO to redefine BCAs as mucinous cystic neoplasm (MCN) or intraductal papillary neoplasm of the bile duct (IPN-B) depending on the presence of ovarian stroma and bile duct communication, respectively [27].
Diagnosis
USG, CT, MRI, magnetic resonance cholangiopancreatography (MRCP), endoscopic retrograde cholangiopancreatography (ERCP) and intraoperative cholangiography have been described for preoperative work up. Classic findings on ultrasound are generally an anechoic mass with internal septations that are highly echogenic. MRI is a valuable tool for the diagnosis and differentiation of cystadenoma from other cystic liver lesions and the combination with MRCP is beneficial [28]. On T1-weighted images, MRI reveals a fluid-containing, multilocular, septated mass with homogeneous low signal intensity, the wall and septa enhancing with contrast injection. Fine needle aspiration (FNA) for diagnostic purposes is not recommended due to risk of dissemination of tumor cells. Elevated levels of serum CA 19-9 and CEA have been reported; however, this is a variable finding. A normal level does not exclude biliary cystadenoma, as some simple liver cysts may also show an elevated serum or cystic fluid CEA or CA 19-9.
Treatment
Due to the significant limitations of preoperative imaging and FNA in differentiating BCA from cystadenocarcinoma, all lesions suspected of being BCA should be surgically removed with a negative margin. Parenchymal sparing resection with occasional ablation of the residual cyst using electrocautery has been reported [29]. Pinson et al. reported cyst enucleation without late recurrence and mortality [30]. This procedure appears to be a valid alternative where resection is difficult or is likely to be associated with significant perioperative morbidity. Good prognosis is observed if total excision is achieved.
Biliary cystadenocarcinoma
Biliary cystadenocarcinoma (BCAC), in contrast to BCA, occurs more frequently in old, male patients and demonstrates a shorter duration of symptoms. Though BCAC and BCA have similar symptoms, differentiation between BCA and BCAC can be made through certain clinical variables that show a significant difference between these lesions in terms of age, sex, bilirubin, ALT, tumor size and presence of mural nodules [30]. Higher enzyme levels in BCAC occur due to the invasion of the adjacent biliary tree, while BCA is a slow-growing tumor and causes late biliary compression. Serum CEA and CA 19-9 are moderate predictors of BCAC [31].
Nakajima et al. divided BCACs into two growth types: invasive (extending into hepatic parenchyma or adjacent organs) and non-invasive (confined to cystic lesion) [32]. Non-invasive lesions have low recurrence and a good long-term outcome. For invasive lesions, the 5-year survival was similar to cholangiocarcinoma at around 22% [31].
Treatment
Radical resection with a wide (> 2 cm) surgical margin should be the goal of treatment. Biliary cystadenocarcinoma has a good prognosis compared to other hepatic malignancies, as it exhibits less aggressive clinical behavior with slow growth and less frequent metastases [33].
A retrospective study conducted by Jwa et al. in South Korea showed that in patients undergoing complete resection there was no tumor recurrence in patients with BCA and in patients with BCAC, 1-, 3- and 5-year disease-free and overall patient survival rates were 100%, 85.7%, 57.1%, and 100%, 100%, 75.0% respectively [34].
Polycystic liver disease
Polycystic liver disease (PCLD) is part of fibropolycystic liver disease manifesting with multiple simple hepatic cysts. It is an autosomal-dominant condition that can be associated with autosomal-dominant polycystic kidney disease (ADPKD), which is found in 50% of these patients. Polycystic liver disease is a rare condition with a slight predominance of females [35]. It is defined by the presence of 20 or more cysts in the liver parenchyma. The prevalence is estimated to be 1 : 158000 people [36]. Prevalence of PCLD is likely to be underestimated as most patients are asymptomatic. 80% of patients have no genetic mutations; however, in 20% of cases mutation in the short arm of chromosome 19 is seen.
Pathophysiology
Pathophysiology of PCLD is heterogeneous; Knudson’s somatic “two-hit hypothesis” may explain this. Individuals carrying germline mutations (first hit) gets the second somatic event (second hit) with secondary loss of function, and these events eliminate the functional gene complex (PC-1 and PC-2), which then modifies the secretory response and initiates focal proliferation and cyst formation. The process involved in cyst growth begins with ductal plate malformation and abnormal biliary cilia, which is associated with cholangiocyte hyperproliferation, fluid secretion, and cyst expansion. There are a few altered intracellular signaling pathways in this disease process involving Ca2+, cAMP and mTOR [37]. The alteration in calcium homeostasis and cAMP activity stimulates activation of mTOR, further augmenting protein kinase-mediated proliferation and cyst growth.
Clinical features
PCLD can cause a significant impact on quality of life. It is more common and more symptomatic in women, as female sex, exogenous estrogen, and multiple pregnancies are risk factors for cyst progression. A minority of patients present with acute complications due to cyst rupture, hemorrhage, infection and compression of adjacent vital structures (inferior vena cava – IVC, portal vein, and biliary tree) [38]. A recent review of 3 randomized, placebo-controlled, clinical trial (RCTs) revealed that the rate of cyst growth is highest in women under the age of 48 [39].
Diagnosis
Even with the marked growth of cysts, liver function tests are usually normal except for a mild elevation in ALP or γ- glutamyl transferase [38]. The most common methods of diagnosis of PCLD are ultrasound and CT due to their widespread availability. On USG liver cysts are well circumscribed and nonenhancing. If there is high attenuation, internal septa bleeding, infection or neoplasm should be considered. MRI is more sensitive and specific, which is useful for preoperative planning [40]. The Gigot criteria are a crude estimation of disease distribution and severity, based on the number and size of the cyst and residual liver parenchyma. The Schnelldorfer classification identifies patients who benefit from resection or transplantation. Treatment should be considered in those cases with persistent symptoms or associated complications [41].
Treatment
There are non-pharmacological and pharmacological treatment options. Nonpharmacological treatment options include cyst aspiration with sclerosis, open or laparoscopic cyst fenestration, combined hepatic resection and fenestration, transarterial embolization and liver transplantation. Pharmacological options include somatostatin analogs (lanreotide and octreotide), mTOR inhibitors, and ursodeoxycholic acid and vasopressin-2 receptor antagonists [2, 42]. Aspiration, combined with ethanol installation, can be effective in patients with a few dominant cysts (Gigot’s type 1 – few large cysts with size greater than 7 cm). Open or laparoscopic cyst fenestration with the omentoplasty approach is another modality of treatment that can be performed in patients with more diffuse PCLD (type 2 – multiple medium cysts 5-7 cm in diameter). Liver transplantation has been performed when the interventions mentioned above were not an option, in patients with concurrent hepatic dysfunction and renal failure, and in patients with diffuse PCLD without segmental sparing. Liver transplantation is associated with 40-50% morbidity and 10-17% mortality [40].
Other cystic lesions of liver
Cystic liver metastases are the most important diagnosis to exclude when multiple cystic lesions are identified in the liver. The common primary sources for cystic hepatic metastasis are colon, kidney, prostate, ovary/testis, squamous cell lung cancer, GIST, sarcomas and neuroendocrine tumors. Chen et al. reported that 50% of all cystic hepatic metastases arise from primary colon cancer [43]. Liver abscess is an important differential diagnosis for cystic hepatic metastases. The cystic nature of the hepatic metastases may be due to development of central necrosis due to more rapid growth of the tumor than its arterial supply. The radiological appearance of the metastases will vary depending on the primary. Clinical history of primary and multiplicity of the lesions are classic features of hepatic metastases. Ultrasound and CT can provide only limited information for differentiation [44].
Undifferentiated embryonal sarcoma (UES) is a highly malignant hepatic neoplasm (typical age at presentation, 6-10 years; and has equal gender distribution); it is rarely seen in late childhood and early adulthood. In the pediatric population, UES is the third most common malignant liver tumor, accounting for approximately 10% of pediatric liver cancers [45]. It is mesenchymal origin with sarcomatous features. Hence some authors suggest that UES arises from mesenchymal hamartoma [46]. Treatment includes surgical resection and multiagent chemotherapy. UES is known to have a very poor prognosis, but emerging reports show a promising prognosis with 20-year survival reported in up to 70% of the patients [47].
Biliary hamartomas, also known as von Meyenburg complexes, are benign congenital lesions consisting of dilated small bile ducts surrounded by fibrous stroma. They are asymptomatic and are almost always discovered incidentally; they require no treatment. Unlike Caroli disease, they do not communicate with the biliary system.
Caroli disease is a benign condition that manifests with saccular dilatation of large intrahepatic bile ducts. Patients who have frequent or recurrent bouts of cholangitis or those with portal hypertension and biliary cirrhosis will benefit from liver transplantation (level of evidence IIc) [1].
Peribiliary cysts are believed to be derived from the periductal glands (extramural-type glands) due to inflammation or deranged portal circulation. They are strongly associated with cirrhosis, portal hypertension, alcohol use, and portal vein thrombosis and autosomal-dominant polycystic disease [1]. Patients are usually asymptomatic, although obstruction of the bile ducts may occur rarely. Hepatic, biliary or pancreatic neoplasms can be associated with peribiliary cysts [48].
An intrahepatic pseudocyst is an infrequent condition that may occur in the setting of pancreatitis, usually as a complication of acute alcoholic pancreatitis [49]. An intrahepatic pseudocyst may require prompt percutaneous or endoscopic drainage or surgical resection if it is large or symptomatic to prevent complications [50].
Biloma is defined as intra- or extra-hepatic bile collection outside the biliary tree with a well-demarcated capsule. It occurs as a result of iatrogenic biliary injury, particularly cholecystectomy, followed by blunt abdominal injury. Most patients with bilomas are treated with nonoperative management. Infected biloma requires surgical intervention such as percutaneous drainage [51].
Intraductal papillary neoplasms of the bile duct (IPNBs) are rare bile duct tumors exhibiting papillary growth within the bile duct lumen. IPNB is considered analogous to the pancreatic intraductal papillary mucinous neoplasm. IPNBs can develop anywhere in the biliary tree, are associated with mucin secretion, and cystic biliary dilatation is characteristic [52]. IPNBs occur commonly in the age group of 50-70 years, and there is a slight male predominance. IPNB should be considered for resection as these lesions can cause recurrent cholangitis and obstructive jaundice even in the absence of malignant transformation. The approach to resection of IPNBs should be similar to resection of intrahepatic cholangiocarcinoma and extrahepatic bile duct carcinomas [2]. Resection generally involves major hepatectomy, segmental resection with regional lymphadenectomy or pancreaticoduodenectomy.
Conclusions
Cystic liver lesions involve a spectrum ranging from benign to premalignant to frankly malignant lesions. Most hepatic cystic lesions are benign and asymptomatic lesions that are diagnosed incidentally and require no intervention. Large, symptomatic or malignant cysts need further evaluation and treatment. Accurate preoperative diagnosis is essential to select appropriate treatment. Premalignant cystic diseases of the liver are rare but pose diagnostic and therapeutic challenges.





