
Introduction
Pheochromocytoma is a rare tumor during childhood [1]. It is a catecholamine secreting tumor that arises from chromaffin cells of the sympathetic nervous system. Its clinical manifestations result from an increasing excretion of catecholamines. The clinical presentation can be variable and assembling many other diseases. The diagnosis benefited from the advancement of imaging, including computed tomography (CT), magnetic resonance imaging (MRI) and metaiodobenzyl guanidine scintigraphy (MIBG). Its therapeutic management is multidisciplinary. Confirmatory diagnosis is based on the histopathological study of the surgical specimen. We report the case of a 7-year-old boy with a multiple and bilateral pheochromocytoma revealed by a short history of abdominal pain and hypertension that has evolved well after surgical treatment.
Case report
A 7-year-old boy with a history of bronchiolitis at the age of 6 months and mumps pancreatitis 2 years ago, has been admitted through the emergency room for a headache, fever, abdominal pain and vomiting evolving for 3 days.
The medical history revealed that the patient had been complaining for more than 2 months of paroxysmal abdominal pain concomitant with headache and sweating for which he received a symptomatic treatment.
At the time of presentation, physical examination showed normal height (121 cm [+0.24 DS]) and weight (18.5 kg [–1.42 DS]), temperature of 38.5°C, a heart rate of 150 bpm, a respiratory rate of 30/min, and a high blood pressure at 138/97 mm Hg, which is considered as a confirmed high blood pressure for his height. The patient was weakly, algic, with profuse sweating. His entire abdomen was tender, particularly in the right lower quadrant, without hepatomegaly, splenomegaly nor palpable mass. The neurological examination showed a Glasgow at 15/15, without meningeal syndrome nor signs of localization.
Biological examinations showed an inflammatory syndrome with a CRP at 194 mg/l, hypokalemia at 3.4 mmol/l, normal glycemia: 5,5 mmol/l, normal natremia: 135mmol/l, and normal renal function.
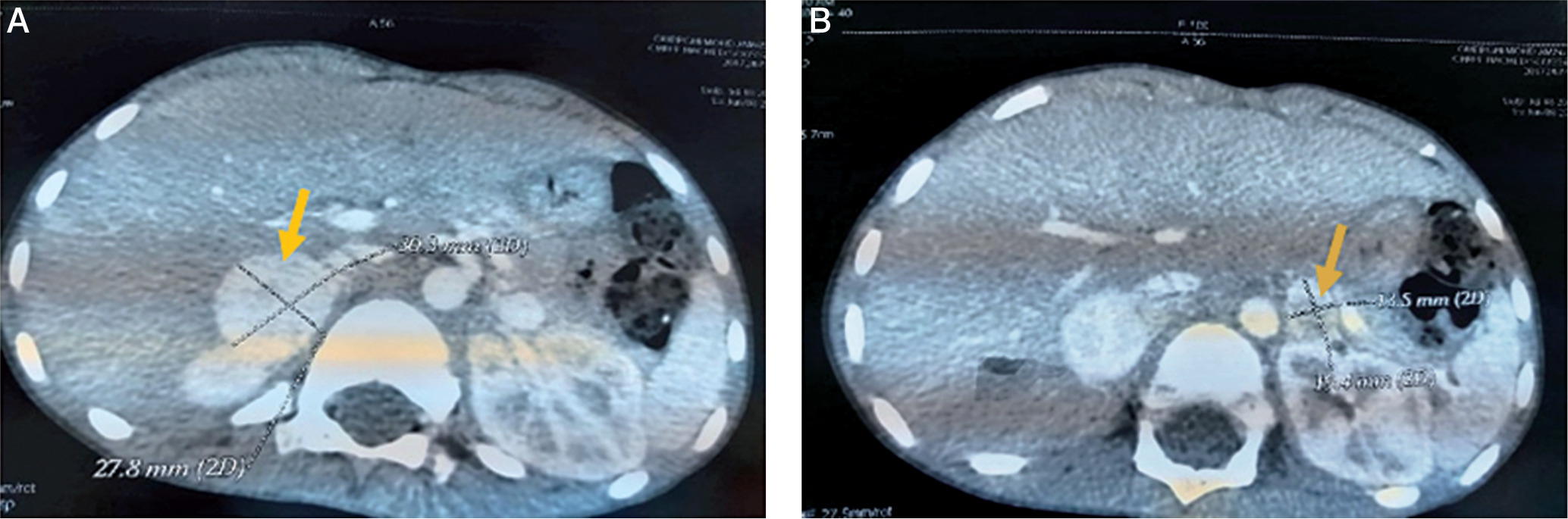
An acute appendicitis was suspected, but the abdominal ultrasound was normal twice. Given the persistence of abdominal pain, vomiting and fever with an altered general state, we completed by an abdominal CT which objectified multiple bilateral adrenal masses: well circumscribed 2 masses measuring respectively 28*26*31 and 27*20*25 mm in the right adrenal gland and 2 other masses measuring respectively 10*10*13 and 17,5*12*16 mm in the left one (Figures 1, 2 and 3). The clinical and radiological presentation was consistent with the diagnosis of pheochromocytoma. To confirm our diagnosis, a methoxy derivative of 24-hour urinary catecholamines dosage was realized, showing an increase in the levels of normetanephrines (realized one day) up to 7.18 mg/24 h (normal range: 0.07–0.46 mg/24 h), and a normal level of metanephrine during 3 consecutive days: 0,16; 0,4 and 0,27 mg/24 h respectively (normal range: 0–1 mg/24 h).
Figure 1
The representative axial images of contrast enhanced CT scan showing mass in the right adrenal gland (A) and left adrenal gland (B)
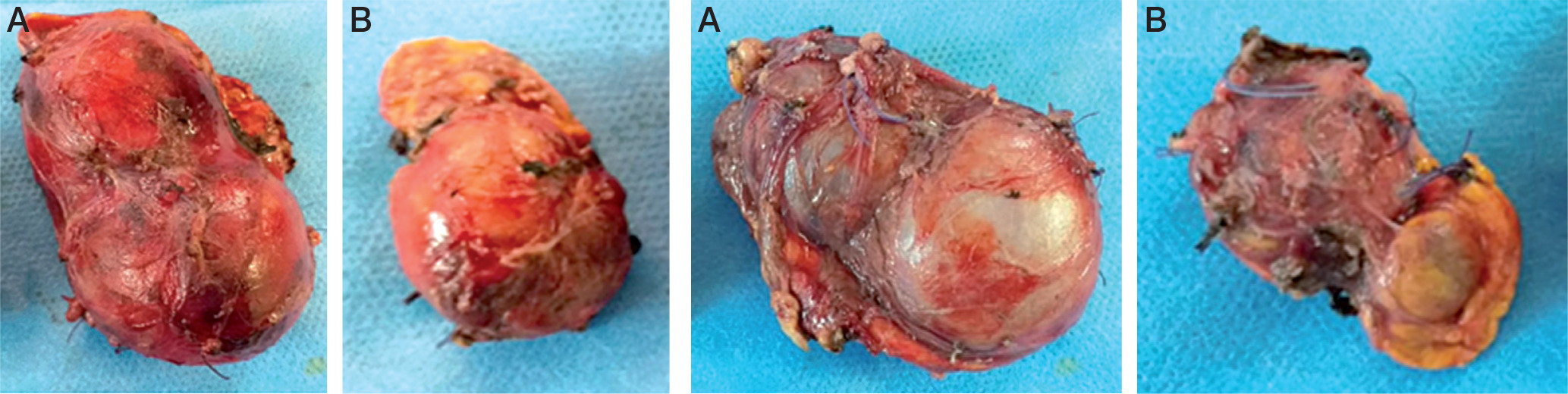
Figures 2 and 3
Total surgical resection of the right adrenal (A). Subtotal adrenal resection on the left side (B). Notice the loss of the normal shape of the adrenals with multi-lobed appearance
The 123I-metaiodobenzylguanidine (123I-MIBG) scintigraphy revealed a bilateral fixation in the right and left adrenals and no other location in the rest of the body.
Screening for other associated disorders that may fit with certain syndromes was carried out: correct calcium-phosphorus balance: calcemia: 2.46 mmol/l, phosphorus: 1.3 mmol/l, PTH: 14 pg/ml (10–65 pg/ml), Prolactin: 171.2 µIU/ml (60–300 µIU/ml), normal plasma cortisol: 65 ng/ml (35–110 ng/ml), thyroid assessment: TSHus: 1.77 mIU/l. Thyroid ultrasound was also normal.
Blood pressure (BP) monitoring showed very high BP figures (systolic BP: 170 to 190 mm Hg and diastolic BP: 100 to 130 mm Hg), requiring the use of intravenous antihypertensive agent (nicardipine). The patient was prostrate and algic most of the time, he had at the time of high blood pressure peaks, an intense headache, abdominal pain and sweating.
The control of the blood pressure was very difficult. We had to associate three antihypertensive drugs to obtain a normal and sustainable blood pressure: calcium channel blockers first intravenously (nicardipine) then orally (nifedipine), a β-blocker (propranolol) and an α-blocker with prolonged release (prazocine).
The blood pressure was controlled after 6 weeks of antihypertensive treatment.
The echocardiography showed a 10 mm hypertrophic interventricular septum with conserved systolic and diastolic function of the ventricules and undilated heart chambers.
Our patient underwent surgery two months after the first consultation; he had a total adrenalectomy on the right side and subtotal on the left side (Figures 2 and 3). During the operation the blood pressure remained balanced without hypertensive peaks during tumor resection.
The pathological examination concluded with multiple and bilateral pheochromocytomas with a Pheochromocytoma of the Adrenal gland Scaled Score (PASS score) between 2 and 6:
the right adrenal gland has 2 tumors measuring 3.5 and 3 cm long axis with a PASS score of 2 (mitosis < 3) and 6 (necrosis, mitosis and spindle cells) respectively,
the left adrenal gland has 2 tumors measuring 2.3 and 1.2 cm with a PASS of 4 (mitosis > 3, atypical mitosis) and 4 (mitosis > 3, marked pleomorphism) respectively,
there was no vascular embolism or capsular invasion.
In the immediate post-operative period, the patient presented severe hypotension (70/35 mm Hg) with hyponatremia (127 mmol/l) related to acute adrenal insufficiency requiring two refills and intravenous hormone replacement therapy: hydrocortisone hemisuccinate 100 mg/m2 and desoxycortone 5 mg/m2, with an oral relay after 2 days by hydrocortisone 12 mg/m2 and fludrocortisone 100 mg per day.
Our patient is now 10 months out from surgery and remains asymptomatic. His examination is unremarkable with correct blood pressure (90/60 mm Hg), good general condition, normal growth and a normal biological balance (natremia 138 mmol/l and kalemia 3,9 mmol/l). An abdominal and pelvic CT scan demonstrated no evidence of recurrent disease.
Family screening for the disease by measuring blood pressure and realizing abdominal ultrasound scan were normal for both parents and brotherhood. Genetic study could not be realized in our patient until now.
Discussion
Pheochromocytoma is a medullo-adrenal tumor developed at the expense of chromaffin cells that secretes epinephrine, norepinephrine, or dopamine. Approximately 98% of patients have abdominal or pelvic tumors, and 2% have thoracic tumors [1]. Commonly associated with hypertensive peaks, it remains a rare case of hypertension and is found in only 0.1 to 0.6% of patients with this condition [2]. Pheochromocytoma is even more exceptional in children, in fact sporadic forms occur generally between 40 to 50 years; and hereditary forms are diagnosed earlier, before 40 years [2]. Despite its low incidence, pheochromocytoma is the most frequent endocrine tumor in childhood [1].
It occurs generally between the age of 6 to 14 years, but early onset in infants is possible even before the age of one year [3].
In children, the familiar forms are more frequent and are often associated with other pathologies, in particular: von Hippel Lindau disease (VHL), von Recklinghausen neurofibromatosis (NF1), as well as multiple endocrine neoplasia (MEN) type 2A and 2B and the familial paragangliomas [2, 4].
The clinical manifestations of pheochromocytoma are polymorphic, therefore it’s referred to as the great mimic [2]. The classic signs including the Menard triad: pulsatile headache, palpitations and profuse sweating, usually dominate the clinic presentation, in addition to pallor and feelings of anxiety and panic. These signs are mainly in relation with the release of catecholamines, but other mechanisms are possible, like hypercalcemia, especially in children [5]. Less common signs and symptoms include fever, nausea, flushing, weight loss and tiredness, and metabolic effects of pheochromocytomas include hyperglycemia and lactic acidosis [2]. Hypertension is classically paroxysmal, with possible hypertensive emergencies. But some patients with pheochromocytoma may present with normal blood pressure (in case of adrenal incidentalomas, or small tumors) or with unexplained orthostatic hypertension in patients with history of hypertension. Pheochromocytoma is the cause of arterial hypertension in 1 to 2% of the pediatric population [2]. Hypertension in children is permanent, and it tends to be more severe than in adults [3]. In a large cohort of 58 patients aged 4 to 20 years with pheochromocytoma, elevation of blood pressure was the most consistent manifestation, and was more likely to be sustained, with or without paroxysms [6].
Another particularity of pheochromocytoma in children is the possibility of other nonspecific clinical signs that confuse more the diagnosis: digestive manifestations (abdominal pain, diarrhea, vomiting) growth retardation, deterioration of the general state, neurological manifestations and convulsions [6]. In our case, the patient presented with an acute surgical abdomen with predominance of abdominal pain and vomiting. However, it is well established that abdominal pain in children have multiple medical and surgical causes, and clinicians should be very careful to ovoid diagnosis traps. During his hospitalization, our patient had a sustained hypertension with severe peaks that were associated with intense headache, pallor, sweating and altered general state. This relation suggests rather extra-adrenal source than adrenal origin of excessive amounts of catecholamines, although we didn’t find extra adrenal tumors in our case.
Once the diagnosis is suspected, patient should undertake specific biochemical exams. Multiple studies showed that the measurements of plasma and 24 h urinary catecholamines (epinephrine, norepinephrine, and dopamine) and urinary vanillylmandelic acid (VMA) had low sensitivity and specificity [7]; and it is now recommended to asses catecholamine metabolites: plasma free metanephrines (metanephrine and normetanephrine) and 24 h urinary fractionated metanephrines [8]. Neverthelsess, the interpretation of their results should remain careful as there are possible false positive in relation with some medications (calcium channel blockers, β-blockers, mood stabilizers…), age, posture, etc., and false négatives in case of small tumors or dopamine secreting tumors [7]. In our observation, the urinary level of metanephrine was normal whereas normetanephrine level was elevated, about 15 times ULN (upper limit of normal). Values > 4 times ULN are highly suggestive of a tumor, whereas values between ULN and 4 ULN request more investigations [9].
Once the biochemical evidence of pheochromocytoma is established, imaging is necessary to localize the tumor. Abdominal ultrasound, often performed as first line, will narrowly miss adrenal tumors due to sensitivity and specificity of 89% and 61%, respectively. Therefore, abdominal-pelvic computed tomography (CT) or magnetic resonance imaging (MRI) are the exams of choice with a sensitivity of 90% to 100% [7]. MRI is superior to CT in localization of extra-adrenal tumors, assessement of the extension into the spinal canal, involvement of major vessels [7], and it is preferred in childhooh or during pregnancy.
As imaging evokes the diagnosis, but remains enable to discern pheochromocytoma from other abdominal pathology, functional imaging studies need to be pursued in order to specify the nature of the tumor and to detect multifocal disease or malignancy. These include 123I-MIBGscan, positron emission tomography (PET) with [18F] fluorodopamine (FDA), [18F] fluorodeoxyglucose (FDG), and [18F] fluorodihydroxyphenylalanine (F-DOPA) [7].
In our patient, abdominal CT and 123I-MIBGscan showed the same finding, without any ectopic localisation.
Surgical management is cumbersome. It requires preoperative preparation in order to control blood pressure and to avoid intraoperative accidents; in fact, tumor mobilization by the surgeon can lead to important catecholaminergic discharges and extreme blood pressure variability [10]. This preparation is mainly based on α- and β-blockers. There has been a drastic decrease in perioperative complications from 45–69 to 3% with the use of α-blockers [7]. Alpha blockade should precede beta blockade in order to offset reflex tachcardia from α-2 receptor antagonism. The preoperative blood pressure goal should be a reduction of < 50 percentile for age and height [7].
Patients are also asked to consume a high sodium diet and fluid intake of at least 1.5 times maintenance a day once on an alpha blocker, to prevent hypotension [7].
In our case, we did not use alpha blockers in the beginning. In fact, as the biochemical investigations were hardly performed, the diagnosis of pheochromocytoma remained hypothetic during weeks, so we used calcic inhibitiors and β-blockers; but this was ineffective to treat hypertension. Blood pressure could be controlled only after adjunction of α-blocker.
As for surgery techniques, laparoscopic resection and adrenal cortical-sparing procedures tend to be preferred, especially in patients with bilateral adrenal disease, to prevent cortisol deficiency. Between 15 and 30% of the adrenal gland is needed to preserve function [11]. Other parameters are important in the decision tree, including risk of disease recurrence and high malignancy rate. Intraoperative risks are mainly hypertensive peaks, which can be controlled by different agents like sodium nitroprusside, esmolol and magnesium sulfate [12]. The resection of such tumors with potential complications require experienced anesthesiologist and surgeon/pediatric surgeon.
Postoperative, prolonged clinical and biological monitoring of patients is required. The long-term evolution of pheochromocytoma in children is marked by the frequency of recurrences, in particular loco-regional and controlateral, and the late onset of metastases [7].
As mentioned above, familiar and inherited forms are more frequent in children. In addition to the known syndromic presentations of MEN II, NF1 and VHL, germline succinate dehydrogensase gene mutations (SDHx) involving the mitochondrial enzyme complex (SDH) form part of the familial pheochromocytomas or paragangliomas [7]. In a series of 164 unrelated pediatric patients diagnosed with pheochromocytoma or paraganglioma, The European-Amercian-Pheochromocytoma-Paraganglioma-Registry (EAPRR) reported that 80% had a germline mutation in a gene associated with such tumors [13]. Lenders et al. proposed an algorithm for genetic testing based on patient’s clinical characteristics, biochemical phenotype and clinical aspects of the tumor [10].
Conclusions
Pheochromocytoma is a very rare disease, especially in the pediatric population. It should always be mentioned in case of severe hypertension in children, but may present with multiple signs and findings. These tumors display important molecular and biochemical differences. The treatment is based on the surgery. Multidisciplinary care (pediatrician, anesthesiologists and surgeons) is essential as well as a long-term postoperative follow-up given the risk of recurrence.









