





INTRODUCTION
The outbreak of COVID-19 in December 2019 in Wuhan, the capital city of Hubei Province in China, and the consecutive increase of the number of infected patients, reaching up to 46 million, has an undoubted influence on various medical and socio-economic issues [1]. Exhaustion of healthcare systems, i.e. overloading of hospital capacity, inability to provide access to all patients requiring respiratory support, and lack of personal protective equipment (PPE), has become a threat even in developed countries with well-established medical care. The pandemic of SARS-CoV-2 infection has also resulted in an unprecedented number of publications regarding various epidemiological, pharmacological, immunological, and medical treatment issues [2]. Considering critical care aspects of COVID-19, the emphasis is mainly placed on respiratory support, ARDS therapy, and prevention and treatment of multiorgan failure caused by dysregulated immune response [3].
What is not sufficiently covered, in the authors’ opinion, is the topic of acute circulatory failure among patients with SARS-CoV-2 infection. Published data, however, suggest that COVID-19 can also affect the cardiovascular system both as a natural consequence of critical disease and as a result of mechanisms unique to the pathogen [4]. From published data, it is evident that the risk of the severe course of the infection and mortality increase with age and the presence of chronic health conditions [5, 6]. Analysis of Chinese, Italian, and US populations reveals that, among comorbidities associated with SARS-CoV-2 severe and fatal course, cardiovascular diseases are one of the most common, with hypertension as the primary one, presenting in 23.0-59.7%, 73.8%, and 56.6-63.0% of cases, respectively [7–11]. The report of The Novel Coronavirus Pneumonia Emergency Response Epidemiology Team that analysed 72,314 COVID-19 patient records reported up to 11 February 2020 revealed that COVID-19 in the presence of cardiovascular disease was associated with 10.5% mortality whereas among patients with coexisting chronic obstructive pulmonary disease mortality was 6.3%, which is only slightly higher than in patients with hypertension [12]. Regarding the presence of haemodynamic instability and shock in COVID-19 among ICU patients, the commentary by Michard et al. states that the proportion of patients in this group, receiving catecholamines, ranges from 35% to 94% in published studies [13]. These data are in line with available information about catecholamine requirement in ARDS patients [14] and suggest that special focus on circulatory failure, and its mechanisms and features in COVID-19 may be beneficial both in understanding the pathogenesis of this disease as well as the proper treatment of critically ill patients with SARS-CoV-2 infection. This article presents the authors’ point of view on haemodynamic failure issues in COVID-19.
MECHANISMS OF CIRCULATORY FAILURE IN COVID-19
There are several potential routes by which the infection causing COVID-19 can affect the circulatory system, both as a primary insult and as secondary to virus-induced pathology (Figure 1). The first mechanism is a direct effect of the viral infection. SARS-CoV-2 is a member of the Coronaviridae family. In its structure, SARS-CoV-2 closely resembles the structure of other known members of this family responsible for previous epidemic outbreaks in the 21st century SARS-CoV (approx. 79% similarity) and MERS-CoV (approx. 50%) [15]. Like SARS-CoV, SARS-CoV-2 uses angiotensin-converting enzyme 2 (ACE2) as a portal of entry into human cells with the promotion of the double-domain glycoprotein present on the surface [S1] of the microbe by the host trans-membrane protease serine 2 (TMPRSS2) [16, 17]. ACE2 receptor has vast expression not only on the epithelial cells of the oral mucosa, airway epithelial cells, and type II pneumocytes, which is probably responsible for the lung pathology in COVID-19, but also in other organs [18, 19]. Its expression was confirmed in the myocardium, microcirculatory pericytes, and different layers of arterial and venous vessels wall [20–22]. Such distribution may be responsible for direct damage to the heart, which can be proven by reported cases of myocarditis in SARS-CoV-2 infected patients, concomitantly with observed vasculopathy leading to vasodilation, thrombotic microangiopathy, and endothelial dysfunction [23, 24]. Interaction of virus particles with ACE2 results in its downregulation [25]. It is of particular importance because the ACE2 plays an important role in counter-regulation of the increased activity of the renin-angiotensin-aldosterone (RAA) system [26, 27]. Reduced density of ACE2 is known for its potential impact on chronic cardiac disease, i.e. increasing the risk of heart failure, whereas overexpression plays a protective role in ischaemic heart pathology [27, 28]. Hence, dysregulation of this system with a possible reduction of the anti-inflammatory effect of angiotensin-(1–7) – a product of the reaction catalysed by ACE2 – may also contribute to circulatory pathology during SARS-CoV-2 infection [29]. However, studies regarding the significance and potential treatment utility of acute effects of those RAA system interactions in conditions like sepsis are sparse. Of note, it is also important that ACE2 receptor plays an important role in animal models in protection from acute respiratory distress syndrome (ARDS) and was suggested as pivotal in the pathomechanism of lung injury by SARS-CoV infection [30, 31].
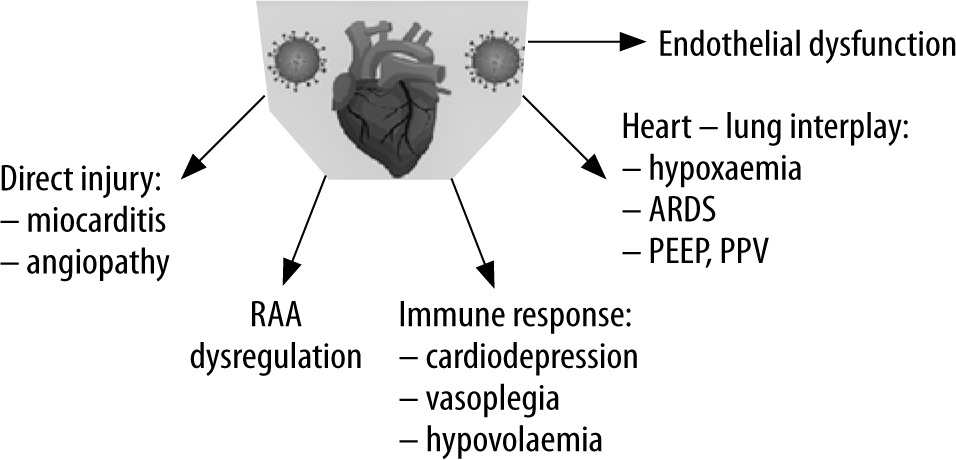
The second mechanism responsible for cardiovascular pathology in COVID-19 is the host’s immunological response to viral infection. In COVID-19 proinflammatory cytokines IL-6, tumour necrosis factor alpha (TNF-α), interleukin 1β (IL-1β), and granulocyte-colony stimulating factor (GM-CSF) are significantly elevated [32, 33]. The Third International Consensus Definitions for Sepsis and Septic Shock defines sepsis as “life-threatening organ dysfunction caused by a dysregulated host response to infection” [34]. This term, typically associated with bacterial or less commonly fungal aetiology, may also be applied to SARS-CoV-2 infection. Hence the term viral sepsis is justified, and its underlying immunological mechanisms may be responsible for symptoms like hypovolaemia, vasoplegia, and cardiodepression [35]. Moreover, approximately 25 to 50% of septic patents develop sepsis-induced myocardial dysfunction (SIMD), leading to various proportions of left ventricular systolic and diastolic dysfunction and right ventricle (RV) impairment [36–38]. The mechanism of this condition is related to the innate immune response by activation of the inflammatory response through toll-like receptors (TLR). Their expression on myocardial cells and interaction with exogenous pathogen-associated molecular patterns (PAMPs) and endogenous damageassociated molecular patterns (DAMPs) that modulate intracellular signalling via, among others, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) that may lead to disruption of contractile proteins [39, 40]. The reaction of TLRs present on monocytes and macrophages increases the production of proinflammatory cytokines, which results in augmentation of the above-described response [36]. The occurrence of SIMD is associated with increased risk of unfavourable outcome [41, 42]. There are no detailed data on its contribution in severe COVID-19; however, it has been diagnosed in other severe viral infections [43].
It is also worth considering that the response induced by SARS-CoV-2 is also being compared to other pathological immune reactions like cytokine release syndrome or secondary haemophagocytic lymphohistiocytosis [44]. Understanding those mechanisms may result in effective therapies modifying the course of the disease.
The third mechanism that may lead to circulatory failure in COVID-19 is associate with endothelial dysfunction. An article by Libby et al. suggests that in severe and in late complicated stages its dysfunction provides a rational explanation for multiorgan failure [45]. SARS-CoV-2 infection, as well as PAMPs, DAMPs, or excessive proinflammatory cytokines, may lead to endothelial layer activation leading, in the severe course of the disease, to coronary plaque rupture, together with alterations of the microvasculature resulting in myocardial ischaemia [45].
The last potential process that may lead to COVID-19-related haemodynamic alterations is secondary to respiratory failure. Cardiovascular and respiratory systems are in dynamic interplay; thus, unsurprisingly, pathologies affecting any one of these systems have a profound impact on the other [46]. From a pathophysiological point of view, the haemodynamic effect of COVID-19-related lung pathology can be mainly attributed to hypoxaemia and consecutive hypoxia, and progressing lung injury (and ARDS) as well as its treatment. Hypoxaemia, which develops in the course of severe COVID-19, causes an increase in respiratory drive and induces changes in pulmonary vascular tone (Euler-Liljastand reflex), which may increase the afterload of the right ventricle and regional vasodilatation in the systemic circulation (cerebral and coronary), along with a general increase in sympathetic drive, resulting in systemic vasoconstriction and tachycardia. The increased respiratory drive may enhance cardiopulmonary interactions during spontaneous breathing. A profound decrease in intrathoracic pressure during vigorous inspiration causes a rise of left ventricle (LV) ejection pressure, LV transmural pressure, and, as a consequence, an increase in afterload and oxygen consumption [52]. Developing lung injury and ARDS may result in the development of pulmonary vascular dysfunction (PVD), which manifests as an increased pulmonary arterial pressure and vascular resistance, potentially leading to severe impairment of RV function [53, 54]. In SARS-CoV-2 infection, the proposed “microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome” (MicroCLOTS) should also be taken into account because it may aggravate the aforementioned pathology [55].
If lungs COVID-19 patent become mechanically ventilated, positive pressure ventilation may diminish systemic venous return, leading to reduction of RV preload and RV stroke volume. Moreover, positive pressure ventilation, high PEEP values and recruitment manoeuvres can increase pulmonary vascular resistance, increasing RV afterload further impeding its function [54].
WHAT DO WE KNOW?
Haemodynamic considerations in SARS-CoV-2 infection should focus on two aspects: adequate monitoring and proper treatment. Unsurprisingly, a new challenge means facing old problems while accommodating for the unique characteristics of the disease. Thus, it is crucial, especially considering the massive number of recently published papers, preprints, and data from more informal sources, to base on recognised knowledge. Such a source may be “Surviving Sepsis Campaign: guidelines on the management of critically ill adults with Coronavirus Disease 2019 (COVID-19)” [56]. Although most recommendations are weak and based on moderate-quality evidence, they provide some baseline knowledge that can be further developed with our rapidly increasing insight into this new disease.
HAEMODYNAMIC MONITORING
Surviving Sepsis Campaign COVID-19 guidelines and a literature review do not suggest that this disease will profoundly affect established principals of haemodynamic assessment, like the preference of dynamic over static parameters. However, it needs to be emphasised that COVID-19 and associated acute cardiovascular failure create a new frontier for cardiovascular monitoring. The utility of precise optimisation of tissue perfusion cannot be overestimated. Nonetheless, in this particular clinical setting, the use of haemodynamic monitoring must be carefully considered due to safety issues.
All invasive procedures in SARS-CoV-2-infected patients must be performed regarding the risk of acquiring the infection by medical staff, with the use of appropriate PPE. Moreover, even a haemodynamic test deemed as minimally invasive or non-invasive, like the passive leg-raise test, may cause a substantial threat, for example by accidental disconnection of an improperly secured breathing circuit. Thus, a careful and structured approach is apt (Figure 2). Consistent with COVID-19 and sepsis guidelines, the authors of this study suggest the initial use of markers of tissue perfusion and hypoxia. The utility of such parameters as capillary refill time (CRT), lactate clearance, CO2 gap, and central venous oxygen saturation (ScvO2) in septic shock was confirmed in numerous studies. The choice of the used parameter should depend on staff experience and local protocols. The use of parameters detecting tissue dysoxia resembles an idea that lies behind the concept of early goal-directed therapy [57]. Although the ProCESS, ARISE, and ProMISe trials do not support the mortality benefit of this approach, it is certain that successful treatment of shock requires reversing pathology occurring at the level of microcirculation [58–61]. Regarding the choice of a parameter, the ANDROMEDA-SHOCK trial did not show a difference in 28-day mortality between the use of either CRT or lactate clearance, recommended by SSC guidelines [62, 63]. It may prove beneficial not to rely on a single parameter because they are relatively easy to obtain. Such an approach may be beneficial in clinical scenarios like normal ScvO2 values in hyperdynamic septic shock with arterial-venous microcirculatory shunting [64]. If changes in cardiac output exert an impact on markers of tissue perfusion and hypoxia, haemodynamic monitoring should be considered in the second stage. The idea of a two-phase approach to haemodynamic monitoring, in the authors’ opinion, would prevent clinicians from unnecessary use of potentially invasive monitoring in situations where haemodynamic manipulations, such as fluid resuscitation, do not impact tissue dysoxia. A review by Monnet et al. indicated that in certain clinical scenarios like early septic shock, there is no utility in confirming an obvious condition, and such assessment may be harmful because it delays necessary fluid therapy [65]. One may assume that a severe COVID-19 patient admitted to ICU with a possible history of severe fever and associated dehydration is indeed fluid responsive. We must take into account that fluid responsiveness is, in fact, a physiological condition [66]. Its assessment is justified only in patients presenting features of circulatory failure where treatment of volume deficit may be beneficial [65]. Inadequate aggressive volume resuscitation, as proved by Maitland et al., may be harmful [67]. In both haemodynamic monitoring and volume therapy, the proper approach should consider situations like concurrent ARDS where restrictive fluid admini-stration is recommended, or the post-resuscitation phase of sepsis and septic shock where large fluid boluses may be of limited utility [68–70].
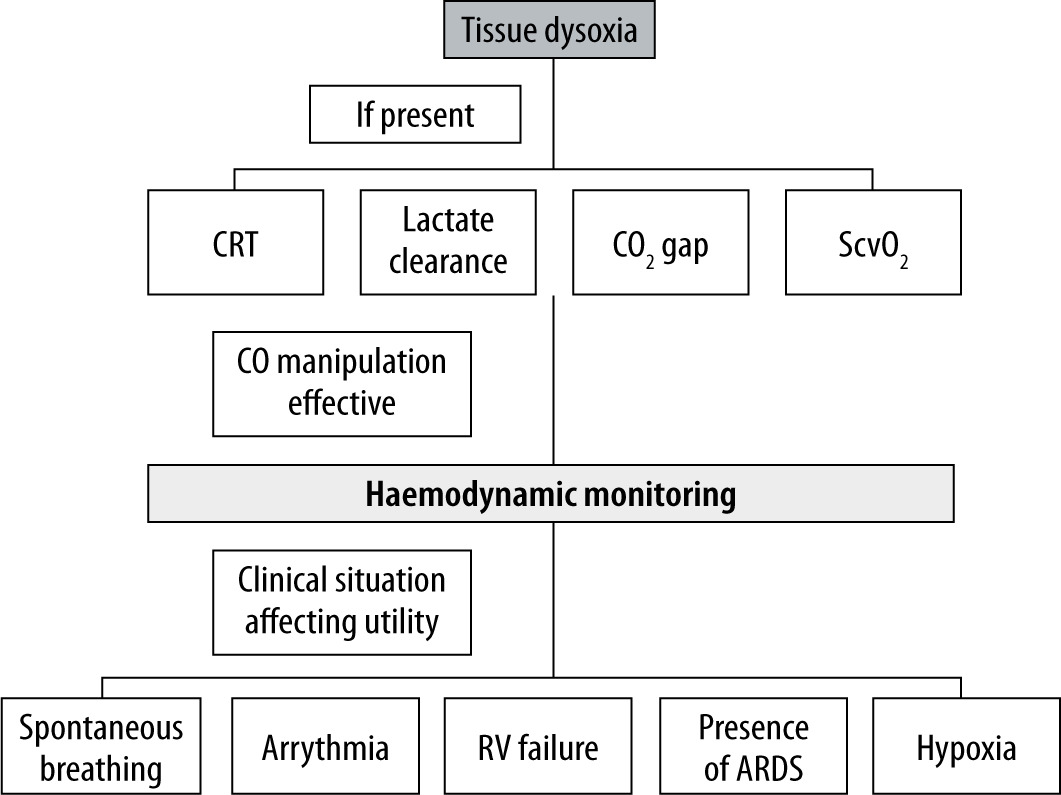
Regarding the choice of haemodynamic monitoring, one must take into account the availability of local protocols and experience with a particular method. The accuracy of cardiac output and stroke volume assessment is crucial when performing a dynamic mini-fluid challenge test. Also, the patients’ specificity must be taken into account. Presence of arrhythmia or a spontaneously breathing patient makes the popular haemodynamic parameters of stroke volume variation (SVV) and pulse pressure variation (PPV) irrelevant. If the right ventricular failure is present, which is likely in ARDS in the course COVID-19, those tests may have a false positive result, increasing the risk of erroneous therapeutic decisions [71]. A false negative result, on the other hand, may occur when the patient has reduced lung compliance caused by ARDS and when a low tidal volume ventilation strategy is being used. Such a condition may be present in “H” type phenotype of SARS-CoV-2 infected lungs, as described by Gattinoni [48]. The inability to perform an inspiratory hold may also reduce the utility of the end-expiratory occlusion test [72]. It is also worth considering that, especially with the use of uncalibrated pulse contour-derived measurements, prone positioning may vastly affect the accuracy of measurements [73].
What can be seen in available reports and is consistent with current knowledge is the use of bedside echocardiography, mainly transthoracic (TTE), for cardiac function assessment and fluid responsiveness assessment. In the results of an international survey by Michard et al. echocardiographic examination was the most common method, with a frequency exceeding 50% [74]. Although TEE methods such as inferior vena cava (IVC) respiratory variations and velocity-time integral (VTI) share limitations, they seem a reasonable choice among other methods in patients undergoing low tidal volume mechanical ventilation, as they are time- and cost-effective as well and easily accessible especially in suboptimal conditions with a limited number of available haemodynamic monitors [74, 75].
Despite haemodynamic optimisation and associated fluid responsiveness, one must bear in mind that both in sepsis-induced ARDS and in COVID-19 restrictive fluid therapy is recommended [56, 63, 68]. However, informal sources, discussions, and opinions presented online suggest a remarkable problem, not adequately addressed in published studies, with the adaptation of this strategy to COVID-19 patients. Patients in severe course of SARS-CoV-2 infection are admitted to the ICU after various durations of fever, hyperventilation, diarrhoea, and fasting. The presence of those symptoms may be responsible for initial severe dehydration. Mechanical ventilation, use of high PEEP values, diuretic use, and persistent fever may aggravate this condition. Our routine assessment of the patient’s fluid status is typically based on fluid charts and body weight measurements. However, those methods have not been proven to be sufficiently accurate and may provide erroneous data for the clinician [76, 77]. This may easily lead to a situation where targeting zero or negative balance leads to severe dehydration and its consequences. Thus, the SARS-CoV-2 pandemic accentuates the need for a structured and objective approach to this subject.
PHARMACOTHERAPY – VASOPRESSORS
Regarding pharmacotherapy of cardiovascular failure due to SARS-CoV-2 infection, SSC COVID-19 guidelines recommend an approach consistent with previous guidelines on septic shock management. Noradrenaline is suggested as a first-line vasoactive agent, and if the target mean arterial pressure cannot be maintained, vasopressin should be considered as a second-line treatment [56]. A review of published data on COVID-19 does not suggest a different approach. However, potential negative aspects of the use of high doses of exogenous catecholamines must be taken into account. High sympathetic activity and increased concentrations of circulating catecholamines are typical in the critically ill and play an important, evolutionary adaptive role, but efforts to overcome myocardial depression and vasoplegia developing in inflammatory shock conditions require supraphysiological levels of both external and internal catecholamines. At some point, this beneficial response loses its evolutionary role of providing, along with the immune system, urgent and adequate reaction to ongoing injury. This state may, as a result, induce unwanted effects related to the suppression of innate and adaptive immunity, bacterial growth and virulence promotion, hypercatabolism, alterations in splanchnic circulation, stress cardiomyopathy, interference of coagulation pathways, etc. [78]. In many conditions such as acute coronary syndromes or traumatic brain injury, catecholamine excess is associated with worse prognosis [79, 80]. High concentrations of circulating catecholamines are also associated with long-term complications of ICU treatment such as ICU-acquired weakness [81].
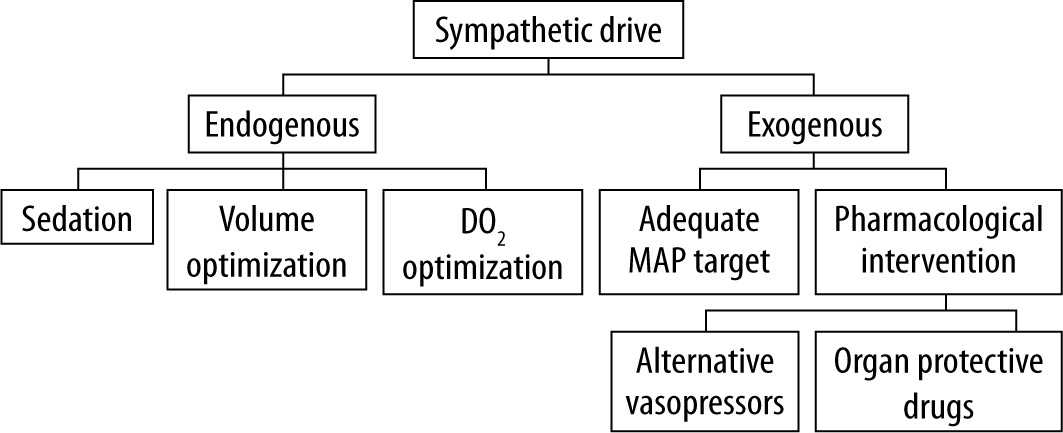
To prevent those potentially detrimental effects of excessive adrenergic stress, the strategy of decatecholaminisation was proposed. This term encompasses measures that are directed at the reduction of both endogenous catecholamine release and exo-genous administration (Figure 3) [82]. Discussion of all aspects of this strategy is beyond the scope of this review; however, in the authors’ opinion, it is crucial to consider potential alternative drugs such as non-catecholamine vasopressors or drugs with potential organoprotective action. Vasopressin was under scrutiny for its potential effect on reducing the risk of acute kidney injury progression in septic patients, which was sparked by post hoc analysis of the VAST trial [83]. Results of the VANISH trial did not confirm such a hypothesis; however, because the study design was probably underpowered, further discussion on this subject is warranted [84, 85]. It is important because COVID-19 patients have a risk of AKI reaching 22.2–36.6% [10, 86].
In 2017 the Federal Drug Administration, and in 2019 the European Medicines Agency, approved angiotensin II (AT II) as a novel vasopressor to treat refractory hypotension in adults with distributive shock. These decisions were based mainly on the results of ATHOS – 3 trial. It is worth emphasising that the results of the trial published by Khanna et al. provide proof that this drug is a potent vasopressor, but the trial does not show improvement in 7- and 28-day mortality, which were additional endpoints of the study [87]. AT II is associated with potentially unwanted effects such as proinflammatory or procoagulant ones [88, 89]. In the setting of severe pulmonary pathology caused by COVID-19 the potential promotion of capillary leak and fibroproliferation, caused by AT II, must also be taken into account [90]. Regarding its use in SARS-CoV-2-infected patients, one must take into consideration that, because of the virus entry receptor (ACE2), this infection may be associated with the potential level of RAA system dysregulation. The addition of external AT II may theoretically lead to unwanted effects resembling a situation with exogenous catecholamine use. As mentioned before, the importance of this fact is not fully established.
In COVID-19 patients, as well as the decatecholaminisation strategy, the choice of proper pharmacotherapy may also take into account the effect of vasopressors on pulmonary circulation (Table 1). Two features of this group of drugs, in the authors’ opinion, should be taken into consideration: changes in pulmonary arterial pressure (PAP) and the influence on hypoxic pulmonary vasoconstriction (HPV). The clinical significance and potential benefit of the effects exerted by particular vasopressor drugs need to be established.
BETA-BLOCKERS
Anticipation of the potentially deleterious effect of sympathetic stimulation leads to the not novel idea of β-adrenergic blockade in sepsis and septic shock as a method of reducing the harm induced by excessive adrenergic stimulation. Studies on animal models show that such therapy may result in improvement of haemodynamic parameters: heart rate, blood pressure, cardiac output, and stroke volume; and reduction of the inflammatory response by decreasing TNF-α and IL-6 concentration, NF-κB activity, and IL-18 expression [99, 100]. In COVID-19, drugs from this group need consideration because in the course of this disease direct and indirect myocardial injury, sympathetic overstimulation, and possible proarrhythmic effects of antiviral therapies may vastly increase the risk of new-onset arrhythmia. Moreover, one of the proposed mechanisms of SIMD is of β-adrenergic dysregulation caused by downregulation of β-adrenergic receptors and disturbance of β-adrenergic signalling [101, 102]. This mechanism underlies the role of dobutamine, a predominantly β-1 receptor stimulating catecholamine, which is recommended for patients with COVID-19 and shock with evidence of cardiac dysfunction and persistent hypoperfusion [56]. While the use of this drug may lead to cardiac output improvement, in the study by Hernandez et al. it did not exert a beneficial effect on microcirculatory perfusion. Infusion of inotropes may, in fact, lead to unfavourable results in terms of mortality [103]. An increase of cardiac index may be associated with an unfavourable change in myocardial oxygen demand [104]. These facts may serve as another potential basis for the β-adrenergic blockade.
Data regarding the use of β-blockers in septic patients are inconclusive. Data from the retrospective analyses on patients treated with β-blockers before admission to the ICU indicate that this therapy may be associated with a mortality benefit. Morelli et al., in a study on septic shock patients with a heart rate above 95 min-1 and high noradrenaline dose, presented a safe reduction in heart rate by infusion of esmolol as well as a possible impact on ICU and hospital mortality. However, in this trial design mortality was not a primary endpoint, and, what is more, its value in the control group was abnormally high – reaching 90% [105]. Recently, a study by Kakihana et al. showed that the ultra short-acting landiolol reduces the incidence of new-onset arrhythmia in septic patients [106]. An important feature of most of the published studies on this topic is that they assess the effect of β-adrenergic blockade by its ability to reduce tachycardia. Such an attitude may not always be appropriate regarding the risk of cardiac decompensation [107]. It may not fully reveal the potential of β-blockers because it is unknown if other postulated mechanisms such as anti-inflammatory, metabolic, etc. are more important in terms of the expected mortality benefit. Results of further trials like STRESS-L, ESMOSEPSIS, and THANE are needed to verify the effects of the sympathetic blockade in this group of patients.
OTHER DRUGS
Another drug suspected of potential organ-protective effect among septic shock, including SARS-CoV-2-infected patents, is dexmedetomidine. This α2 receptor agonist, which revolutionised sedation in ICUs, in animal models reveals a possible anti-inflammatory and organ-protective impact [108]. The results of the DESIRE trial did not show improvement in mortality or ventilator-free days in septic patients receiving this drug [109]. However, in the subgroup with severe sepsis (APACHE II ≥ 23) dexmedetomidine use was associated with lower mortality. Recently published post hoc subgroup analysis of the trial by Nakashima et al. suggest an effect on improving renal function [110].
Levosimendan, a calcium sensitiser with inotropic and vasodilatatory properties, effective in heart failure treatment, has also been shown to exert pleiotropic effects on other organs. As a result, its use may beneficially impact haemodynamic variables, microcirculation, kidney, and liver function. Those effects, along with the antioxidative antiapoptotic and anti-inflammatory properties of levosimendan, suggest the role of this drug in the treatment of septic patients. Unfortunately, while early studies suggested mortality benefit, the LEOpards trial, a large RCT, did not confirm less severe organ dysfunction or lower mortality in septic shock patients treated with levosimendan [111–113]. In this study levosimendan administration was an addition to standard care in a prespecified, albeit small, subgroup of patients with a low cardiac index. Thus, this study might have not reliably excluded benefits in specified subgroups.
Although the idea of a pharmacological approach to decatecholaminisation seems promising, it has, as well as the controversies mentioned above, one major drawback regarding the effects of each drug. As mentioned in the commentary by Chawla et al. regarding vasopressors, there is no test available at the bedside that can assess which combination of vasopressors and organ protective drugs is beneficial for particular patients [114]. Moreover, even for vasopressin, the use of which is well established, there are no recommendations as to what point it should be added or how its combination with noradrenaline should be weaned.
It must also be stressed that so far, no evidence allows us to universally adopt the idea of decatecholaminisation. As mentioned before, published data on COVID-19 do not suggest a different attitude than this presented in SSC COVID-19 and previous guidelines. The time at which the number of patients presenting with COVID-19 shock often exceeds the capacity of our ICUs is not a suitable moment to change our established practice on that matter. Finding answers and solutions to all questions and caveats associated with the concept of decatecholaminisation, use of alternative drugs, and the clinical significance of the various effects of vasopressors on pulmonary circulation will undoubtedly exceed the time course of the SARS-CoV-2 pandemic.
CONCLUSIONS
SARS-CoV-2 infection has a predominantly pulmonary pathology and can vastly affect the cardiovascular system. It is, however, of great importance that, regarding haemodynamic aspects, this viral infection emphasises existing problems with cardiovascular support in the critically ill. Nonetheless, the specific effects of this pathogen on haemodynamics should also be taken into consideration. In the authors’ opinion, it should direct us to evolution rather than revolution regarding our current practice. In countries that were most impacted by COVID-19, many efforts were focused on enabling proper respiratory support outside ICUs. Not only in scientific sources but also in social media pictures of people with CPAP helmets were numerous. Maybe it is also a proper time to reconsider the topic of cardiovascular support, in particular how and to what extent it should be used beyond our ICUs.