
Introduction
The pituitary stalk interruption syndrome (PSIS) is diagnosed based on a characteristic pituitary picture in a magnetic resonance imaging (MRI), showing an interrupted or thinned stalk, hypoplasia, or aplasia of the anterior lobe, as well as lack or incorrect location of the posterior pituitary lobe [1]. The pituitary stalk interruption syndrome is one of the complex forms of congenital pituitary insufficiency [2]. To date, the cause of PSIS has not been established, which may result from a wide variety of clinical presentations. As a rule, mutations in genes for signalling molecules and transcription factors associated with early stages of pituitary development cause the symptoms of hypopituitarism in association with extra-pituitary disorders [3-5]. A genetic cause of PSIS is identified in only approximately 5% of all cases [6]. Symptoms resulting from insufficiency of the pituitary gland, despite the inborn character of the disease, may appear at various stages of life. The age of establishing the correct diagnosis depends not only on the severity of hormonal disorders, but also, due to a remarkably diverse clinical manifestation of the disease, from the vigilance of physicians. Most often, endocrinological diagnostics starts in the childhood due to inhibition of growth. In newborns and infants, the symptoms of hypopituitarism are often non-specific. Hypoglycaemia presenting with tremors, apnoea, convulsions, hypotonia may be a red flag. Also, cholestatic jaundice may be caused by ACTH deficiency [7]. Cryptorchidism and/or microphallus may be associated with gonadotrophin deficiency. The function of the posterior pituitary lobe in patients with PSIS is usually preserved [8]. A wide spectrum of hormonal deficiencies is observed in laboratory tests. Insufficiency of pituitary gland can affect one or several hormonal axes (isolated vs. combined pituitary hormone deficiency, CPHD) [1].
Aim of the study
We retrospectively analysed clinical manifestations in 31 patients (15 girls and 16 boys) with PSIS, aged from 1.4 to 16.2 years (median 5.7 years), hospitalized in the Department of Paediatric Endocrinology and Diabetology from 2008 to 2020 due to hypopituitarism. In all our patients (100%) the anterior pituitary lobe was visible. In 11 children (35.5%), complete agenesis of the stalk was found, and hypoplasia of the pituitary stalk was found in the remaining 20 children (64.5%). The ectopic posterior pituitary lobe was diagnosed in 28 subjects (90.3%), while in 2 patients the posterior lobe was not visible, and in the following 1 child it was residual (9.75%). In 15 children (48.4%) features of septo-optic dysplasia (SOD) were found.
Material and methods
All children were examined by the same team of Department’s physicians. The patients were weighed on electronic scales with an accuracy of 0.05 kg, the height was assessed using the Harpenden stadiometer with an accuracy of 0.1 cm. For each child body mass index (BMI) [kg/m2] was calculated. Standard deviation scores (SDS) for BMI and height were calculated [9]. The growth velocity and the puberty (Tanner stages) were analysed. Blood for the laboratory examinations was collected in accordance with the principles of good practice from the ulnar vein. The hormonal function of the pituitary gland was assessed at the beginning of endocrine diagnostics, at median age of patients 5.1 years (range 0.2–16.1 years) and during regular control visits.
In short-statured children who were more than two years old, night growth hormone (GH) profile (screening test after falling asleep) and two GH stimulation tests (with glucagon [dose 0.03 mg/kg body mass], and with clonidine [dose 150 µg/m2 body surface] or arginine [dose: 0.5 g/kg body mass, maximum dose 30 g]) were performed. In patients with normal growth, only the night GH profile was established. GH concentration was measured using chemiluminescence method with a Siemens reagent (cat. no. L2KGRH2). Insulin-like growth factor-1 (IGF1) was determined in all children [10]. The thyrotropic axis was evaluated based on simultaneous analysis of TSH and FT4 concentrations [11]. The corticotropic axis was evaluated based on morning ACTH and cortisol concentrations. If the cortisol concentration was within the range of 3–13 µg/dl, a stimulation test was performed after administration of tetracosactid and/or glucagon (one or both tests were done) [12-14]. The gonadotropic axis was evaluated in girls after 13 years of age and in boys after 14 years of age without puberty signs [15]. The initial assessment of posterior pituitary function was based on the analysis of the fluid balance. In the case of polyuria, the diagnostics was extended to the water deprivation-vasopressin test [16]. In all patients, magnetic resonance imaging (MRI) of the hypothalamic-pituitary region was performed.
Growth hormone deficiency (GHD) was diagnosed if the peak GH response to stimuli was below 10 ng/ml in all performed tests. In short-statured children younger than 2 years old GHD was diagnosed according to hypothalamic-pituitary defect (PSIS) in the MRI and deficiency of at least one additional pituitary hormone [10]. The criterium for diagnosing secondary hypothyroidism was the presence of an inadequate TSH values in relation to low FT4 levels at least two times [11]. Secondary adrenal insufficiency was diagnosed when the ACTH concentration was in the lower normal range and the morning cortisol concentration was below 3 µg/dl or when the cortisol concentration did not exceed 18 µg/dl after tetracosactid administration, or 14.6 µg/dl in the glucagon test [12, 13, 17]. Hypogonadotropic hypogonadism was diagnosed when delayed puberty in physical examination was present, prepubertal concentrations of estradiol/testosterone were found, as well as no increase in gonadotropins concentrations after GnRH analogue administration [15]. Diabetes insipidus was diagnosed in accordance with the standards when in the serum osmolality exceeded 300 mOsm/kg H2O with simultaneous urine osmolality below 300 mOsm/kg H2O in the water deprivation test. If urine osmolality was within the range of 300–750 mOsm/kg H2O, partial diabetes insipidus was diagnosed [16]. CPHD was diagnosed when at least two pituitary trophic hormones axes were disturbed. Data were expressed as percentages and as median and ranges.
Results
In 17 patients (54.8%) the main reason of the referral to the endocrinology department was short stature, including 3 children with abnormal results of thyroid hormone tests and 1 child with PSIS (MRI was performed before hormonal diagnostics due to the optic nerve hypoplasia). Two children with normal height and SOD in MRI of the brain were admitted due to suspicion of secondary hypothyroidism. Four children were referred to the clinic due to hypoglycaemia. One 2-year-old boy was admitted after severe hypoglycaemia, which occurred after the first dose of L-thyroxine given due to secondary hypothyroidism. Other reasons for admission were: growth deficiency and delayed sexual maturation (1), suspected Cushing’s syndrome (1), microphallus and secondary hypothyroidism (1), short stature and diabetes insipidus (1), subclinical hypothyroidism and assessment of adrenal function during chronic steroid therapy due to drug-resistant epilepsy (1), SOD in MRI scan and secondary hypothyroidism and adrenal insufficiency in laboratory test results (1), continuation of the treatment of CPHD diagnosed in the 3rd day of life in the other centre (1).
Anamnesis
Twenty-six children (83.9%) were born on time, 5 (16.1%) prematurely (34–37 Hbd). The low birth weight was found in 2 newborns. Eighteen children (58%) were born via Caesarean section (CS), including three due to the breech position. Additional children's diseases are presented in Table 1. In Patient No. 3 in the 6th day of life, a craniocerebral trauma occurred because of a traffic accident. In the neonatal period and infancy, hypoglycaemia was observed in 13 subjects (41.9%). Thirteen newborns (41.9%) were treated for infection. After the birth, cryptorchidism was found in 2/16 and microphallus in 3/16 boys. In 1 girl on the 4th day of life an exchange blood transfusion was performed due to hyperbilirubinemia. One boy underwent surgery due to gastroschisis, in the postoperative period complicated by sepsis. In 9 children (29%), the course of the neonatal period was normal. Two infants (6.5%) had cholestatic jaundice. In 5 subjects (16.1%) hypoglycaemia was observed during childhood, in 2 children it occurred during full hormonal supplementation of CPHD. Optic nerve hypoplasia was observed in 13 subjects (41.9%). Epilepsy was diagnosed in 5 patients (16.1%).
Table I
Clinical characteristics of children with PSIS
Anthropometrics and hormonal evaluation of the studied children
At the beginning of endocrinological diagnosis of patients the median height standard deviation score (HtSDS) was –3.13 (from –7.31 to 1.35), while BMI SDS was 0,02 (from –1.96 to 7.09). Obesity was diagnosed in 5 children (16.1%), while short stature was found in 26 patients (83.9%). Only 5 children (16.1%) had normal growth, including 3 infants (patients No. 4, No. 6, and No. 8) and 2 children at the age of 8.8 year (patient No. 18) and 4.4 year (patient No. 19). In the follow-up, however, four of them showed inhibition of height velocity and consequently – growth deficiency.
In currently 13-year-old patient (No.18) with TSH deficiency (diagnosed at the age of 7.8 years) and ACTH deficiency (diagnosed one year later), short stature was found for the first time when she was 12.9 years old (HtSDS –2.72, BMI SDS 3.02). During her endocrine follow-up, at the age of 10.7, low GH concentration (< 0.05 ng/ml) in the night GH profile, as well as low IGF-1 concentration (25.9 ng/ml, normal range: 88–452) were demonstrated. The diagnostics of GHD (including stimulation tests), was not extended due to the inability to treat GH as part of the therapeutic program of a patient with a normal height (at that time, HtSDS - 0.83, BMI SDS 4.14). Finally, at 12.9 years of age, GHD was confirmed (maximum GH surge in stimulation tests < 0.05 ng/ml), together with celiac disease (tissue transglutaminase antibodies IgA > 200 RU/ml, normal range: < 20 RU/ml; endomysium IgA antibody titre 1:1000, negative titre: < 1:10).
The height of 1 boy (patient No. 6) with ACTH deficiency (diagnosed at the age of 0.4 year) and TSH deficiency (diagnosed at the age of 3.1), currently aged 7.5 years still is within normal range; however, his nutritional status is excessive (HtSDS 0.74, BMI SDS 2.49, duration of endocrine follow-up is 7.4 years).
Two children with initially short stature were confirmed with GHD and fulfilled inclusion criteria for the growth hormone therapy within reimbursed therapeutic program. As a result of the parents’ decision, growth hormone therapy was either not started or discontinued. During further follow-up, however, the children achieved normal height (patients No. 1 and No. 11).
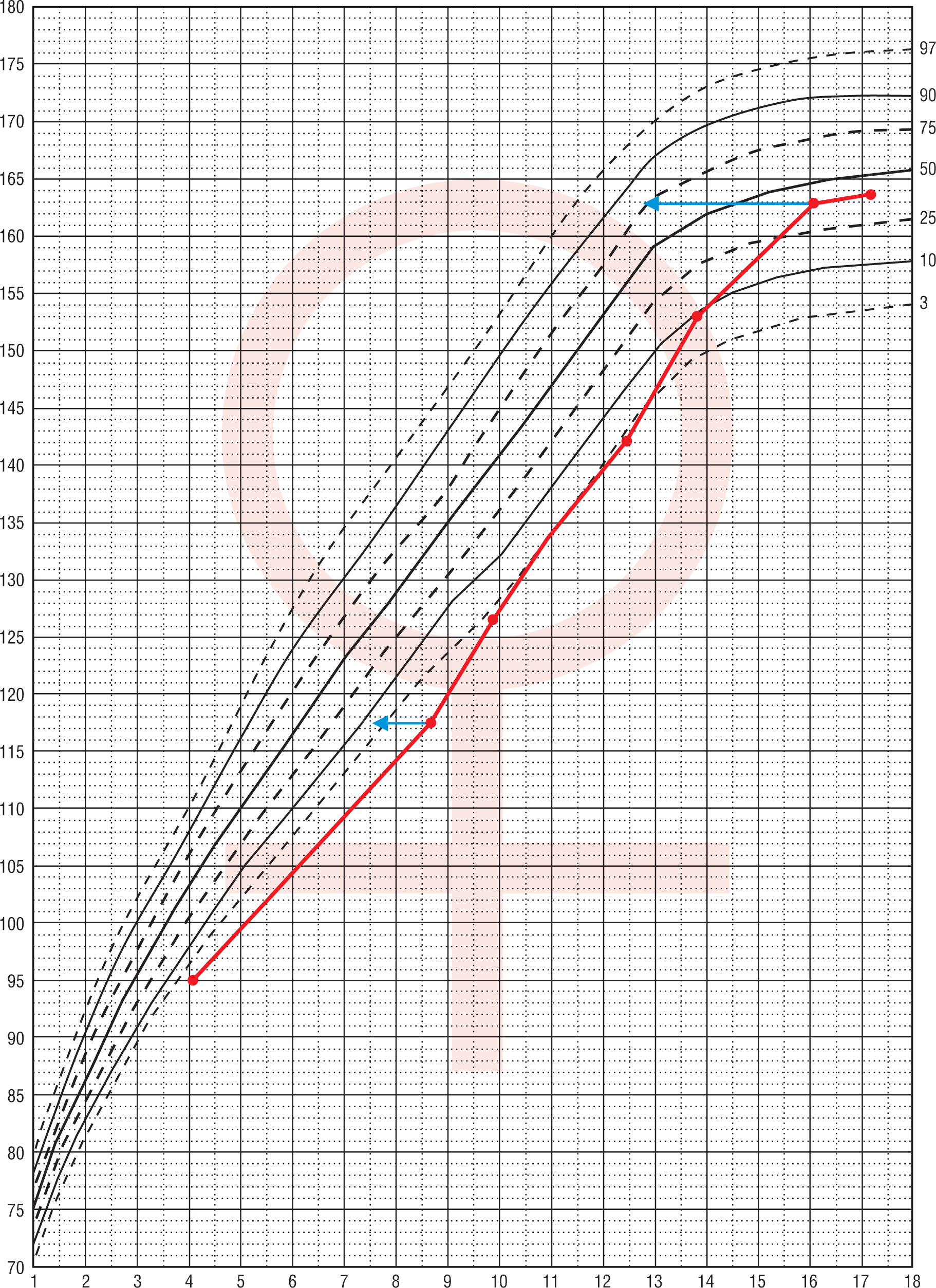
Patient No. 1 was hospitalized for the first time at the age of 4.1 years due to symptomatic hypoglycaemia (HtSDS –2.58, BMI SDS –0.03). Hormonal evaluation then and repeated at the age of 8.7 (HtSDS –2.88, BMI SDS 0.45) revealed low IGF-1 and GH levels in stimulation tests (respectively maximal GH 1.54 ng/ml and 0.26 ng/ml). Growth hormone treatment was not undertaken because of parental denial. From 12.4 years she was treated for secondary hypothyroidism. At 16.1 years of age, the patient presented with primary amenorrhea. On admission, normal height, and nutritional status (163 cm, HtSDS –0.32, BMI SDS 0.54) was found, with pubertal maturation of 1° according to Tanner scale. Her respective target height is 157.5 cm. Periodically, the patient experienced episodes of marked weakness and dehydration, especially during an infection. Hypogonadotropic hypogonadism was diagnosed and secondary adrenal insufficiency (cortisol at 8:00 a.m. 0.6 µg/dl, ACTH 11.1 pg/ml) was found. Despite L-thyroxine (50 µg/day) substitution, thyroid function was decompensated (FT4 9.16 pmol/l, normal range: 10.3–24.4; TSH 0.317 µIU/ml, normal range: 0.4–4). In oral glucose tolerance test (OGTT), hyperinsulinemia (in 120 insulin 141 µIU/ml) was present, HOMA-IR 2.35, IGF-1 49.3 ng/ml, normal range: 226–903, IGFBP-3: 1.38 µg/ml, normal range: 3.5–9.5). Bone age was delayed by 3 years. Short stature was observed in the patient from 4 to about 10 years of age (no data about height status before the patient was 4), followed by marked acceleration of growth from 12.4 to 16 years of age. Later, after the correction of hormonal disorders at the age 16.1 (adrenal, thyroid, gonadotropic axis) there was a marked slowdown in the height velocity (Fig. 1).
Figure 1
Change of height during life in patient No. 1 The arrows indicate the position in the growth percentiles by bone age
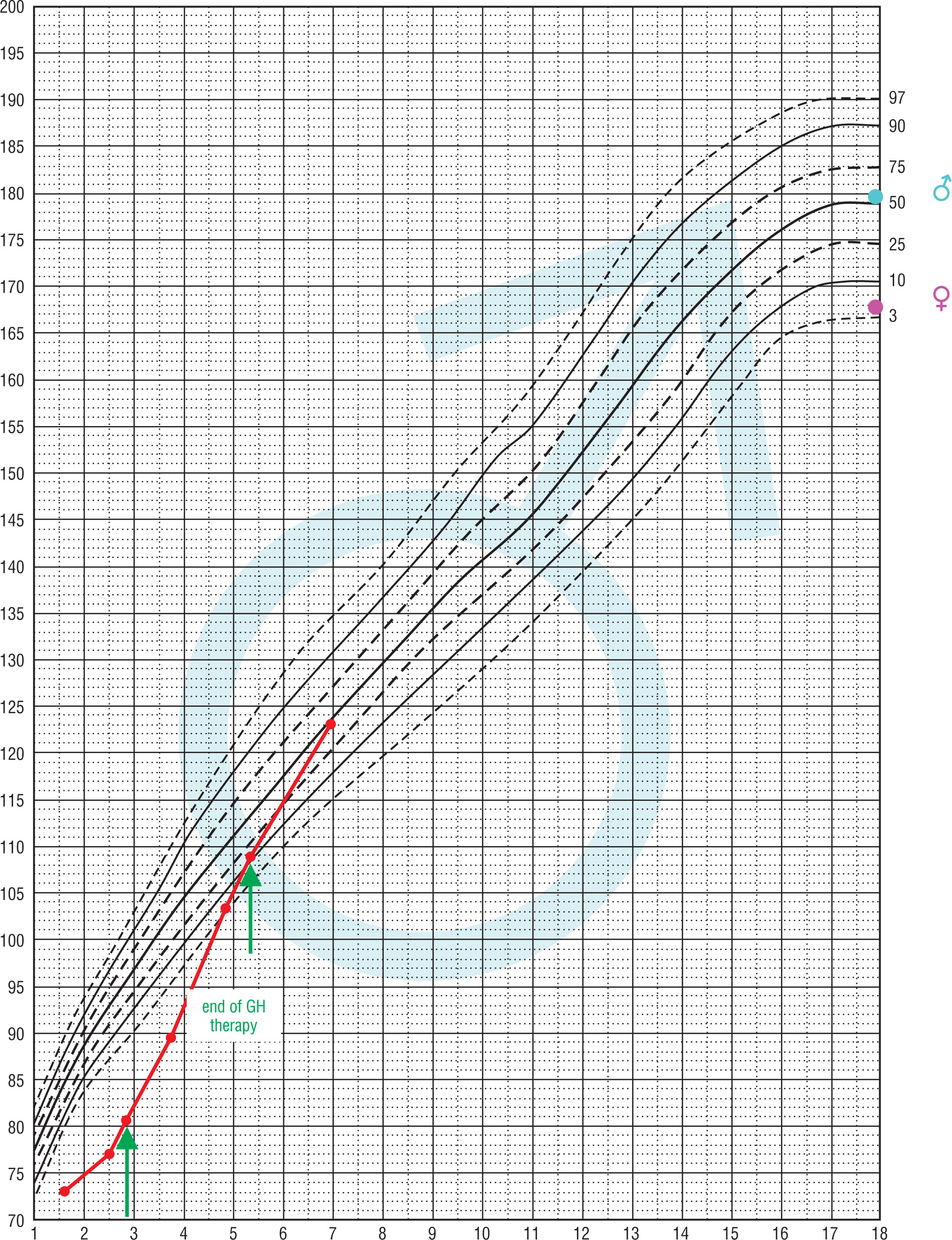
In a currently 7.2-year-old patient (No. 11) with ACTH and TSH deficiency (diagnosed at the age of 1.6 years), GHD was diagnosed at 2.5 years of age. At that time HtSDS was –4.5, BMI SDS 0.77, maximum GH surge in stimulation tests was 0.529 ng/ml, IGF-1 < 25 ng/ml, normal range: 49–289 and IGFBP-3 < 0.5 µg/ml, normal range: 0.9–4.3. Thyroid and adrenal functions were balanced. Initially, he received GH irregularly, then at the age of approximately 5.4 years, the treatment was completely discontinued (HtSDS –1.15, BMI SDS 3.02). From 6.5 years of age, he also did not receive hydrocortisone and L-thyroxine due to parental neglect. At the age of 7, after the intervention of a social worker, the boy reported for medical examination. He presented in a general good condition, with normal height and obesity (HtSDS –0.1, BMI SDS 3.6). Height velocity in the previous year was 8.75 cm/year (without GH therapy). Thyroid and adrenal function were both decompensated (at 8:00 a.m. cortisol 1.7 µg/dl, FT4 5.8 pmol/l, normal range: 11–17.9, TSH 2.6 µIU/ml, normal range: 0.35–5.7; IGF-1 < 15 ng/ml, normal range: 57–316, IGFBP-3 1.05 µg/ml, normal range: 1.4–6.1). Bone age was delayed by 3.5 years. The course of this patient's growth is presented in Fig. 2.
Figure 2
Change of height during life and rhGH treatment in patient No. 11 The arrows indicate the position in the growth in the begining and end of GH therapy
GHD was diagnosed in 30 children (96.8%), including 28 (93.3%) with severe GH deficiency (maximum GH surge in stimulation tests < 5 ng/ml). The median maximum peak of GH in the night profile and stimulation tests was 1.25 ng/ml (range from < 0.05 to 7.95 ng/ml). In one child (patient No. 6) the concentrations of fasting IGF-1 and maximum peak of GH in the night profile were low (evaluated at 5.5 years of age) with maximal GH 0.091 ng/ml, IGF-1 21.1 ng/ml, normal range: 50-286. However, due to the normal body height and growth rate, stimulation tests were not performed. Up to now, secondary hypothyroidism was diagnosed in 20/31 patients (64.5%). Secondary adrenal insufficiency was diagnosed in 21/31 children (67.7%). In 10 patients, adrenal insufficiency was diagnosed based on low morning cortisol concentrations, while in following 10 patients with low cortisol levels a stimulation test was performed to confirm the diagnosis. One of presented girl with adrenal insufficiency was diagnosed in the 3rd day of life in the other clinic abroad (we do not have the initial result of lab test). Among all patients with adrenal insufficiency, 19 patients require permanent substitution therapy with hydrocortisone, whereas the following two, only in stressful situations. Three patients (9.7%) were diagnosed with diabetes insipidus, however in one of the girls in a partial form.
In the whole study population during first examination 8/31 children (25.8%) were diagnosed with CPHD. During the endocrinological observation (median follow-up 5.1 years, range 0.5–13.2) of the above-mentioned group 23/31 subjects (74.2%) were diagnosed with CPHD (median follow-up time 5.2 years, range 0.7–13.2 years). While 8/31 patients (25.8%) were diagnosed with isolated GHD (median follow-up 4.6 years, range 0.5–6.3) (Table II).
Discussion
PSIS is a complex form of congenital pituitary insufficiency. It is diagnosed based on the characteristic radiological picture of the pituitary gland. This syndrome is associated with other congenital disorders such as optic nerve hypoplasia as well as epilepsy or intellectual disability [1, 6]. In the group of studied children, the gender distribution is equal, as it was observed by Pinto et al. [18] and Pham et al. [19]. In some of the described populations of patients with PSIS, boys and men predominate (from 62.7% to 84.7%) [20–23], which may be related to the fact that boys more often report to the endocrinologist because of short stature rather than to X-linked inheritance.
The initiation of hormonal therapy does not always closely reflect the time of a specific hormone deficiency, because the clinical course is often discrete and the beginning difficult to determine. Additionally, physiological interactions between individual hormonal axes should be considered: with cortisol deficiency, there is a spurious increase in TSH, which may delay the diagnosis of secondary hypothyroidism. In turn, thyroxine increases the metabolism of cortisol and therefore the initiation of treatment with L-thyroxine without prior exclusion of cortisol deficiency may result in adrenal crisis and severe hypoglycaemia, which occurred in one of the presented patients.
Analysing the cause of hospitalization, we found that the main reason for referral to the endocrinological department was short stature and the most frequently diagnosed hormonal deficiency was GHD. Our observation is consistent with El Chehadeh et al. [24], who assessed 14 patients with PSIS, revealing that severe GHD (86% of the group) is characteristic for those patients. The age of establishing the correct diagnosis depends not only on the severity of hormonal disorders, but also on the vigilance of physicians. In infancy, hypoglycaemia is the major clinical manifestation of CPHD. Bar et al. [22] compared patients with PSIS depending on the primary manifestation of the disease (n = 67). The diagnosis in the neonatal period was due to the occurrence of hypoglycaemia, while later it was due to short stature or the presence of midline defects. In the group of patients referred to the Department due to short stature, based on a retrospective assessment of available medical documentation, the authors of the study identified symptoms of hypopituitarism in the neonatal period in as many as 14/47 patients (30%). The above observations are consistent with our results, as we have found that hypoglycaemia in the infancy was present in 34.6% of patients (9/26), who were referred to the endocrinologist during childhood due to short stature.
Moreover, attention should be drawn to the diverse clinical course of patients with PSIS. Children differ in the degree of height deficiency as well as the maximum peak GH secretion and the moment of hormonal deficiency presentation. Early growth patterns differ from severe early delay to normal growth [25]. It must be emphasized that not only GHD may be responsible for growth failure. An example of a patient diagnosed with an additional non-endocrine cause of growth retardation is girl No. 18, with GHD and celiac disease.
In the described group, 1 obese patient with normal height at the age of 7 and low concentrations of IGF-1 together with low GH secretion during the night, still require in-depth diagnostics. However in children with obesity GH secretion may be reduced to the level observed in children with classic GHD, which does not necessarily translate into problems with growth. Nevertheless, we take into consideration that further diagnosis of GHD might be needed in the future. The phenomenon of normal growth in patients with confirmed growth hormone deficiency has been observed, although it is not fully understood. They seems to be similar to the case of a 16-year-old girl with PSIS and CPHD described by Lee et al., who despite significant GH deficiency in the insulin test achieved the correct final height – 168 cm [26]. It is inquiring why in a patient with confirmed severe GHD periods with the inhibition of height velocity followed by a normal growth rate are observed, as in presented subjects No.1 and 11. Turkish researchers described similar course of growth in a case of a 9-year-old girl with panhypopituitarism after the neurosurgical procedure due to the craniopharyngioma: after 3 years of slow height velocity she was short statured (HtSDS –4.1), but then she started to gain weight and grown well (HtSDS –1.35). Scientists highlight that there are several hypotheses that explain the phenomenon of growth without GH and evaluating each single case would give insight into the mechanism [27]. Hypothetical reasons for this may include the complexity of hormonal interactions in people with multi-hormonal hypopituitarism, as well as delayed advancement of bone age [28]. In both patients with GHD in our group in the period of accelerated growth, ACTH and TSH axis were not compensated, as well as gonadotropin axis in the adolescent girl. Hyperinsulinemia is another mechanism thought to be responsible for this phenomenon, as insulin receptors are highly homological with IGF-1 receptors and are capable to suppress serum IGFBP-3 resulting in the increase in plasma levels of free IGF-1 [27]. However, Lazar et al. described the growth pattern of 5 children in CPHD who attained a normal final height. They were referred for endocrine evaluation because of a microphallus or delayed puberty. Repeated tests revealed GHD. Patients were not obese. They had no evidence of hyperinsulinemia and unlike our 2 patients, L-thyroxine and hydrocortisone were administered appropriately [29].
Another question is whether early GH replacement therapy is indicated in patients with normal growth, despite GHD? A study by Stewart et al. aimed at assessing the effects of early GH therapy not only on growth, but also on body composition and serum lipids in young children with optic nerve hypoplasia and GHD and normal growth. The results suggested that nearly all children studied ultimately require growth hormone treatment due to growth failure, while the results regarding positive body composition changes as well as lipids were inconclusive [25].
In conclusion, it should be emphasized that PSIS, despite the congenital nature of the disease, may occur at diffrent stages of life, as diverse signs and symptoms of hypopituarism are. It is therefore up to the viligance of physicians to ensure that the correct diagnosis is made as early as possible even though the symptoms of hypopituarism would not be obvious.
Conclusions
Children with PSIS present a diverse clinical picture and should be carefully observed because of the risk of further pituitary disorders.
In the differential diagnosis of hypoglycaemia in the neonatal period and in infancy, hypopituitarism should be considered. In the group of children with pituitary hormone deficiency, especially CPHD, hypoglycaemia may be a serious clinical problem.
The phenomenon of normal growth in patients with confirmed GHD has been observed, although is not fully understood.









