Introduction
Central diabetes insipidus, currently defined as arginine vasopressin deficiency (AVD), is a heterogeneous group of disorders characterized by polyuria–excretion of large amounts of hypoosmotic urine, polydipsia, hypernatraemia, and hyperosmolality of serum. Due to the causes of impaired synthesis and/or secretion of antidiuretic hormone (arginine vasopressin peptide – AVP), 2 types of AVD can be distinguished: congenital and acquired. Congenital forms of AVD most commonly occur in association with congenital brain abnormalities such as: septo-optic dysplasia, holoprosencephaly, and congenital hypopituitarism [1–3]. Although over 80 mutations have been identified in the AVP gene locus, familial forms account for only 10% of AVD cases. Among acquired forms of AVD, the most common causes are central nervous system tumours (embryonal tumours, histiocytosis, craniopharyngioma), head injuries and neurosurgical procedures, and inflammatory and autoimmune processes. The prevalence of AVD is 1 in 25,000 in the population, with the vast majority being acquired forms; AVD symptoms develop when more than 80% of AVP-secreting neurons are damaged [1–6]. The diversity of causes poses a diagnostic challenge for endocrinologists because excluding expansive processes is a priority in this group of patients. Recent reports present detailed diagnostic schemes that allow for rapid and effective differential diagnosis of expansive and inflammatory processes in patients with AVD. However, specific recommendations for long-term, comprehensive care for children with AVD to improve their quality of life during development and in adulthood have not yet been defined.
The aim of this study is to develop a precise diagnostic and therapeutic strategy to improve the quality of life of patients with AVD during development and adult life.
Material and methods
A retrospective, 25-year analysis of imaging changes, hormonal disorders, and auxological changes in children dia- gnosed and treated for AVD from 1998 to 2023 at the Department of Paediatric and Adolescent Endocrinology Children’s Hospital in Cracow was performed. Thirty-one children diagnosed with AVD were analysed: 21 boys (68%) and 10 girls (32%). The distribution of the analysed data was assessed using the Kolmogorov-Smirnov test. Calculations were performed in Statistica 14 and Excel 365. The average follow-up period was 61 months (range: 1–222 months), 20 patients completed the follow-up (including 2 who died), and 11 children continued the follow-up (marked *, Tables I–V). The duration of the disease was assessed from the first symptoms to the diagnosis of AVD and to determination of the aetiology, on the basis of which the children were divided into 8 groups (Tables I–V). Diabetes mellitus, hypercalcaemia, hypokalaemia, and chronic kidney disease were excluded as causes of polyuria in all patients. Diagnosis was made based on clinical and biochemical symptoms: polyuria of over 2 l/m2/day, or over 150 ml/kg/day in newborns, 100–110 ml/kg/day up to 2 years of age, and 40–50 ml/kg/day in older children; polydipsia; urine specific gravity < 1,003 g/l; serum Na > 145 mmol/l; serum osmolality > 300 mOsm/kg H2O; and urine osmolality < 300 mOsm/kg H2O. Dehydration and vasopressin tests were performed in 21 patients. Among the 14 patients treated surgically, AVD occurred before the procedure in 7 children. In the remaining 7 patients after neurosurgical treatment, a 3-phase course was observed: polyuric phase due to the inhibition of ADH release, inadequate vasopressin secretion (SIADH), and AVD due to depletion of ADH stores. In 3 children with early-onset AVD due to congenital disorders of the central nervous system, a dehydration test was not performed. Growth hormone (GH) deficiency was diagnosed by showing GH concentration < 10 ng/ml in 3 stimulation tests: 2 pharmacological ones (after the administration of clonidine, glucagon, or arginine) and an overnight test. The overnight test was conducted until 2020. In recent years, 2 pharmacological tests were performed in accordance with the guidelines. Secondary hypothyroidism was diagnosed based on low circulating free thyroxine (FT4) concentration associated with low or normal serum TSH (FT4; free thyroxine N: 10–25 pmol/l; TSH thyrotropic hormone N: 0.3–4.0 uIU/ml), and secondary adrenal insufficiency was diagnosed based on reduced cortisol (N: 50–230 ng/ml; 137.95–634.57 nmol/l) and ACTH (adrenocorticotropic hormone N: 10–60 pg/ml) concentrations at 8 a.m. or after the administration of 1 µg of synacthen. Sodium concentration (N: 134–143 mmol/l) in serum was assessed using the potentiometric method on a Vitros 4600 analyser.
Table I
Detailed analysis of the patients with AVD according to the etiology - idiopathic
Table II
Detailed analysis of the patients with AVD according to the etiology - brain tumors
ACTHD - adrenocorticotropic hormone deficiency; AFP - a-fetoproteine level; AVD - arginine vasopressin deficiency; p-HCG - human chorionic gonadotropin; BMI - body mass index; GH - growth hormone; GHD - growth hormone deficiency; HbA1C - glycated hemoglobin test; m - months; No - patient's number; PPBS - posterior pituitary bright spot; TSHD - thyroid-stimulating hormone deficiency; y - years
Table III
Detailed analysis of the patients with AVD according to the etiology - post-traumatic and hemorrhagic
[i] ACTHD - adrenocorticotropic hormone deficiency; AVD - arginine vasopressin deficiency; AVP - arginine vasopressin peptide; BMI - body mass index; GH - growth hormone; GHD - growth hormone deficiency; m - months; No - patient's number; PPBS - posterior pituitary bright spot; y - years * patient during follow up
Table IV
Detailed analysis of the patients with AVD according to the etiology - congenital central nervous system malformations
[i] AVD - arginine vasopressin deficiency; BMI - body mass index; GHD - growth hormone deficiency; TSHD - thyroid-stimulating hormone deficiency, ACTHD - adrenocorticotropic hormone deficiency; GH - growth hormone; HCT - hydrocortisone; m - months; No - patient's number; y - years * patient during follow up
Table V
Detailed analysis of the patients with AVD according to the etiology - unfinished diagnostic processes
Urine and serum osmolality (N: 50–1200 and 275–305 mOsm/kg H2O, respectively) were measured using the temperature-dependent method on an 800 CLG Trident-med osmometer. Hormonal assays, luteinising hormone (LH; N: < 1.0–3.7 in prepubertal children; 1.0–18.54 mIU/ml in follicular phase; 1.08–8.34 mIU/ml in boys), follicle-stimulating hormone (FSH; N: < 1.0 mIU/ml in prepubertal children, 2.53–8.00 mIU/ml in follicular phase; 3.0–15.4 mIU/ml in boys), testosterone (N: < 1.0 in prepubertal boys; 2.59–10.95 ng/ml in pubertal boys), oestradiol (N: < 7 pg/ml in females up to 8 years of age; 8.2–17.8 pg/ml: between 8 and 12 years of age; 16.0–34 between 12 and 14 years of age; 10.1–77.6 in follicular phase after 14 years of age) were performed using the radioimmunoassay method on a Siemens Centaur XPT analyser. Insulin-like growth factor (IGF1) levels (N: values depending on age and gender are given in the Tables I–V) were determined using the immunochemical method with a radioactive label (Dia Source). Chorionic gonadotropin β (β-HCG; N: < 5 mIU/ml) and α-fetoprotein (AFP; N: < 10.0 ng/ml) concentrations were determined using the immunochemical method using direct chemiluminescence on an Adria Centaur XPT device (Siemens). Magnetic resonance imaging (MRI) of the pituitary- hypothalamic region with gadolinium contrast was performed in all patients in AP and sagittal projections, and images in T1 and T2 projections were analysed. The MRI examination was performed using GE HDX Echospeed 81.5 T and GE Optima 450 W, 1.5 T machines. Densitometric assessment of the lumbar spine L2–L4 was conducted with a Hologic Horizon Wi device (S/N 300257M). Z-score BMD values were then calculated, taking into account the bone age of the patients (N: Z-score from +1 SD to –1 SD). A retrospective analysis of the aetiology of AVD, as well as auxological, hormonal, and bone mineral density assessments were performed.
Results
The median age at the onset of AVD symptoms was 9 years and 9 months (from 1 month to 17 years), while the median age at AVD diagnosis was 10 years and 2 months (from 1 month to 17 years and 11 months). The average time of the onset of polydipsia and polyuria to AVD diagnosis was 10 months (from 1 to 86 months), whereas the average time from AVD diagnosis to aetiology determination was 14 months (from 0 to 86 months). In 28 patients, the initial symptoms were polydipsia and polyuria. In 3 children, dehydration, high serum sodium concentrations, and lack of thirst due to damage to osmoreceptors in the hypothalamus (adipsic diabetes insipidus) were observed. In 4 children, hemiparesis was observed; in 5 patients, facial nerve palsy; in 5 patients, visual disturbances; in 2 patients, anisocoria; in 3 children, blindness; in one child, cerebral palsy; in one child, quadriplegia; in 3 patients, epilepsy; in one patient, convergent strabismus; in one patient, Horner’s syndrome; and in 3 children, headaches. One female patient experienced excessive sleepiness.
Upon admission to the hospital, the median urine specific gravity in the analysed patient group was 1.0016 g/l (1.001–1.0006 g/l). The median serum osmolality was 302.14 mOsm/kg H2O (280–341), and the median urine osmolality was 207.29 mOsm/kg H2O (37–556 mOsm/kg H2O). The sodium concentration averaged 145.3 mmol/l (131–159.2 mmol/l). All patients received desmopressin treatment with monitoring of fluid balance, urine specific gravity, body weight, and serum sodium concentration. A basic assessment of the anterior pituitary function was performed in all children. Out of the 31 analysed children, idiopathic AVD was diagnosed in 3 children (9.7%; group 1, Table I). In 19 cases (61%), the cause of AVD was central nervous system tumours (Table II): in 12 (38,7%) AVD occurred before the diagnosis of an expansive process, and in 7 (22.6%) after neurosurgical treatment. The most common causes of AVD were germinoma in 5 patients (26.3%, group 3) and histiocytosis in 4 patients (21%, group 2). Among the 6 patients with craniopharyngioma (31.5%, group 5), 2 developed AVD before surgery. In the remaining 4 patients, AVD was a consequence of neurosurgery, as was the case with 3 (15.7%) children diagnosed with pilocytic astrocytoma (group 4). One patient was diagnosed with pituitary adenoma (incidentaloma) as the cause of AVD (5.2%).
In 3 children (9.7%), a central nervous system malformation was identified as the cause of AVD; while in 2 (6.5%), it was due to head trauma, and in 2 (6.5%), due to haemorrhage or hydrocephalus (Table III). One patient with a normal pituitary appearance was diagnosed with familial AVD (3.2%), while another boy undergoing oncological diagnostics was suspected of having an inflammatory central nervous system condition (3.2%).
Auxological assessment
Age of symptom onset: the oldest in our observation was a patient with AVD with suspected lymphocytic hypophysitis – 16 years and 4 months, and patients after astrocytoma surgery – aged 16 years and 3 months. The median age for children with craniopharyngioma was 11 years and 8 months. Patients with post-traumatic AVD, pituitary adenoma, germinoma, and histiocytosis were of similar age (median age 10 years 9 months, 10 years 8 months, 10 years 8 months, and 10 years 6 months, respectively). The median age for patients with post-traumatic AVD is 10 years 7 months, for those with post-haemorrhagic AVD it is 12 years, and in the patient diagnosed with genetically determined AVD, symptoms appeared at the age of 7 years 2 months. The youngest group with AVD is patients with congenital CNS malformations – the median age at diagnosis was one year.
In all 31 patients the median height at diagnosis was (–) 0.64 SDS (from –5.8 to 3.31 SDS), and at the end of observation (–) 1.09 SDS (from –10.33 to 2.7 SDS), Δ SDS –0.43 (from –8.3 to –5.09). At the time of diagnosis of AVD 8 patients had short stature (≤ third centile), median (–) 2.97 SDS, which worsened to (–) 4.61 SDS at the end of observation. During the observation period: 15 patients’ growth slowed down, this applies primarily to all patients (100%) with craniopharyngioma and congenital CNS malformations (100%; Figs. 1–2).
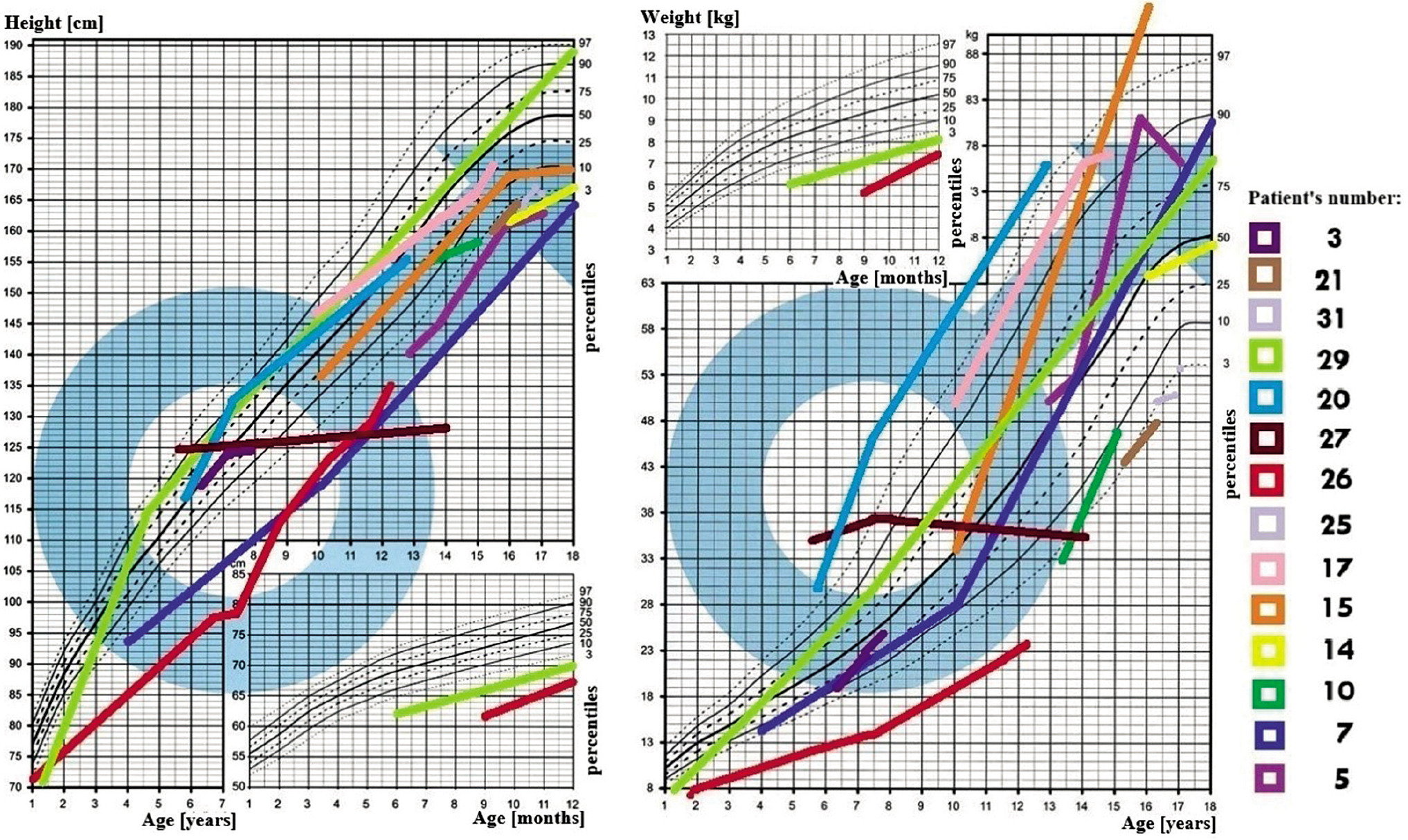
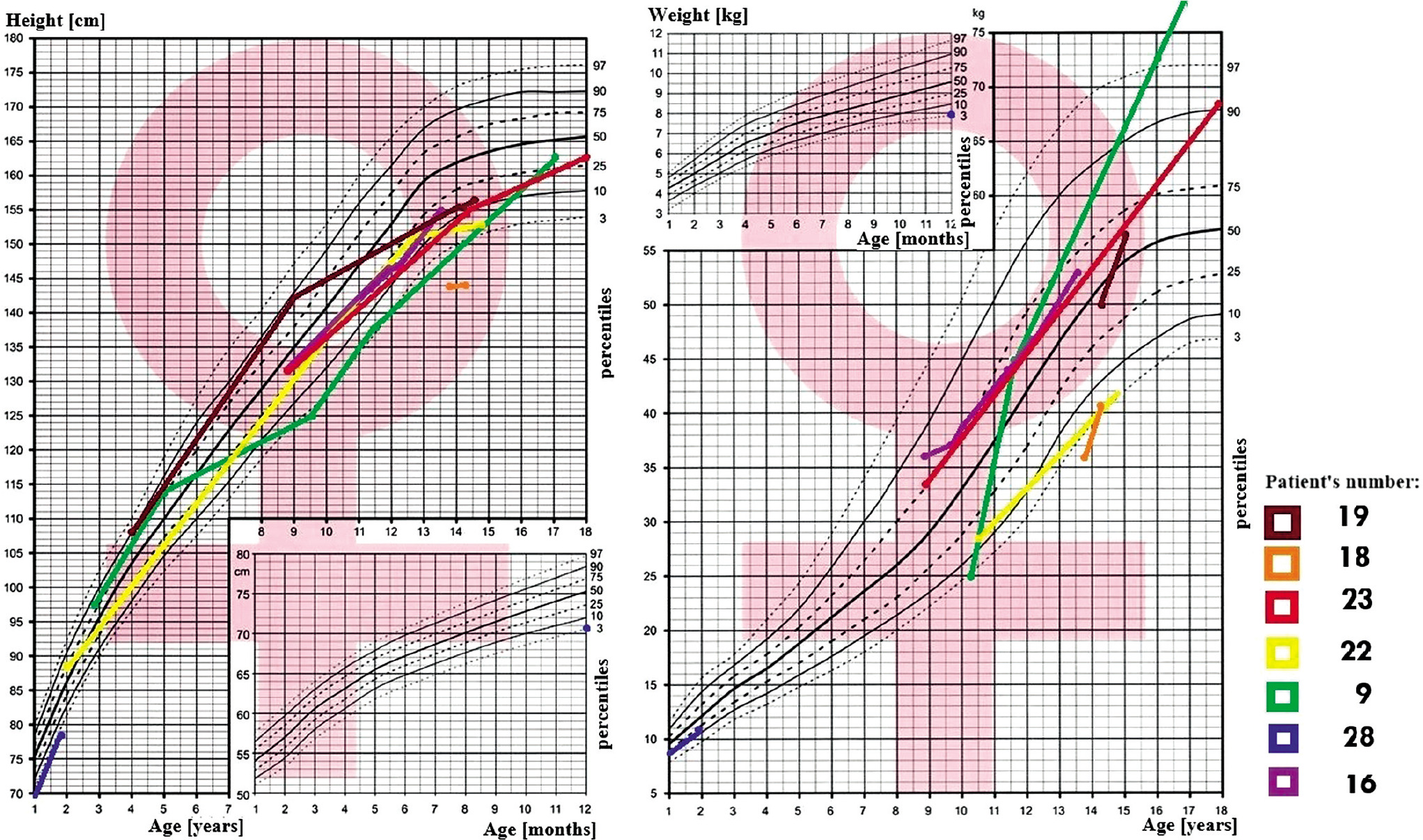
The BMI value in all 31 patients at the time of diagnosis was on average 0.75 SDS (range from –1.97 to 9.58 SDS), and at the end of observation it was 1.16 SDS, Δ BMI SDS 0.23 (range from –8.95 to –2.68). At the time of diagnosis of AVD, 11 patients were overweight or obese; the BMI in this group was on average 3.25 SDS. During follow-up, a total of 13 patients with AVD were found to be overweight or obese. An oral glucose tolerance test was performed in 6 patients, revealing insulin resistance in 5 of them and type 2 diabetes in one.
Hormonal assessment
GH deficiency was found in 9 patients, GH stimulation tests were performed in 8 out of 31 patients revealing in 7 of them a maximum GH concentration < 10 ng/ml. In 2 infants with multihormonal pituitary insufficiency, GH deficiency was diagnosed based on the occurrence of hypoglycaemia and auxological data. Central hypothyroidism was diagnosed in 12 patients; secondary adrenal insufficiency in 11; transient adrenal insufficiency in one patient with post-traumatic AVD; and hypogonadotropic hypogonadism in 5 (Tables I–V).
Assessment of bone mineral density
Densitometric tests were performed in 6 patients, showing low bone mass in 5 of them (Table VI). The mean Z-score for bone age was –1.68 SD (range: from 1.1 to –3.2 SD). Low bone mass was found in 5 out of 6 examined children. The lowest Z-score values were found in children after craniopharyngioma treatment (patients nos. 16, 17, and 18). In patient no. 3, with normal bone mineral density, the examination was performed before the initiation of treatment for LCH. No bone fractures were reported in the patients (Table VI).
Table VI
Bone mineral density in patients with AVD
Analysis of groups separated by aetiology
In group 1 (Table I), 3 patients with a diagnosis of idiopathic AVD remain under observation. In this group, the only abnormality observed was the absence of the characteristic signal from the posterior pituitary lobe. Growth deficiency or obesity was not observed in these patients. The average growth rate in this group was Δ SDS –0.14 (range: from –0.53 to 0.29 SDS), Δ BMI –0.8 SDS (range: from –1.96 to 0.1 SDS), and the mean observation time was 77.3 months (range: 14–118 months). In a 25-year retrospective analysis of 31 children, of whom 6 were initially diagnosed with idiopathic AVD, 2 turned out to suffer from germinoma with a long delay (diagnosed after 5 and 2 years of AVD symptoms, individually). Only 1 of 6 patients diagnosed with idiopathic AVD completed follow-up at 100 months without additional hormonal or ophthalmic symptoms.
In group 2 of children diagnosed with Langerhans cell histiocytosis (LCH) (Table II), 2 patients had a disseminated form and 2 had a pituitary focus. In this group, 2 patients suffered from short stature or growth retardation and obesity (67%). The average growth rate in this group was Δ (–) 2.19 SDS (range: from –7.71 to –0.02 SDS), Δ BMI SDS 0.72 (range: from –0.99 to –2.68 SDS), and the mean observation time was 78 months (range: 9–169 months). GH stimulation tests were performed in both boys, confirming GH deficiency in patient no. 7, who did not receive recombinant human growth hormone (rhGH) treatment due to recurrence of LCH.
Thickening of the pituitary stalk and a deceleration in growth rate were also observed in patients in group 3 with pituitary germinomas (Table II). In this group, AVD preceded the diagnosis of tumours in all cases, with an average time to establish the aetiology of 28 months (ranging from 1 to 60 months), often due to infrequent imaging examinations. In patients no. 8 and 9 with an initial diagnosis of idiopathic AVD, germinoma was diagnosed after 60 and 24 months, respectively. The average growth rate in this group of patients was Δ SDS (–) 0.08 (range: from –2.41 to 3.18 SDS), Δ BMI 0.64 SDS (range: from –0.64 to 2.32 SDS) median observation time 65.8 months (from 1 to 103 months). Obesity was observed in 2 patients (40%). Two patients (40%) had elevated tumour markers in their serum.
In group 4 patients with astrocytomas (Table II), AVD occurred postoperatively. Growth retardation was observed in 2 patients, and GH secretion was not assessed in any of the patients. The growth rate in this group averaged Δ SDS –0.63 (range: from –0.24 to 0.63 SDS), Δ BMI: 1.41 SDS (range from –0.36 to 2.68 SDS), median observation time: 10.3 months (3–16 months).
In 60% of group 5 patients with craniopharyngiomas (Table II), AVD symptoms occurred postoperatively. All (100%) had growth deficiency/retardation, mean Δ SDS –0.49 (range: from –1.65 to 0.16 SDS), Δ BMI: 0.15 SDS (from –0.81 to 1.22 SDS), median of observation time 39 months (range: 2–96 months). All patients (100%) had multihormonal hypopituitarism, and 3 (50%) had obesity. Of the 6 patients after craniopharyngioma surgery, GH secretion was assessed in 3 patients, revealing pituitary somatotropin hypofunction – treatment was initiated. Patient no. 20 with multihormonal hypopituitarism and hypothalamic obesity after surgery died as a result of thromboembolic complications.
In group 6 patients (Table III) with post-traumatic and post-haemorrhagic AVD, growth deficiency/reduction was observed in 3 patients (75%), mean Δ SDS 0.53 (range: from –0.92 to 2.77 SDS), Δ BMI: 0.63 SDS (range: 0.52–0.95 SDS), median observation time 69.25 months (1–151 months). Patient no. 23 was diagnosed with GH-deficiency (the girl discontinued treatment), and boy no. 24 had transient secondary adrenal insufficiency. Patient no. 26, with post-haemorrhagic AVD, was dia- gnosed with GH deficiency – treatment was initiated.
In group 7 children (Table IV) with congenital CNS malformations, AVD symptoms required early diagnosis and treatment. All patients had growth retardation and multihormonal hypopituitarism (100%). For growth, the average Δ SDS: –0.97 (range: from –8.3 to –5.09 SDS), Δ BMI: –2.17 SDS (range: from –8.95 to 1.68 SDS), mean observation time 117.3 months (range: 17–222 months). In patient no. 27 with septo-ophthalmic dysplasia, hypernatraemia was misdiagnosed and treated from birth: hydrochlorothiazide and furosemide were used. He was referred to our clinic at the age of 5 years with significant psychomotor delay. In this boy AVD (adipsic type), hypothyroidism, and adrenal gland insufficiency were diagnosed – treatment with desmopressin, thyroxine, and hydrocortisone was started. Although the IGF1 concentration was below the norm for age, dynamic tests were not performed due to the mother’s lack of consent for further diagnostics. The child died from an infection.
Group 8 (Table V) includes 3 children with incomplete diagnosis. In a girl with a pituitary adenoma, apart from AVD, no dysfunction of the anterior pituitary gland was detected until adulthood. The patient with familial AVD was discharged without genetic diagnosis, the diagnosis was made only on the basis of family history (the father of the patient with AVD). In this group, 3 patients (75%) had growth retardation, mean Δ SDS: 0.36 (range: from –1.06 to 1.97 SDS), Δ BMI: 0.50 SDS (range: from 0.05 to 1.37 SDS), median observation time 45.6 months (range: 2–86 months).
Discussion
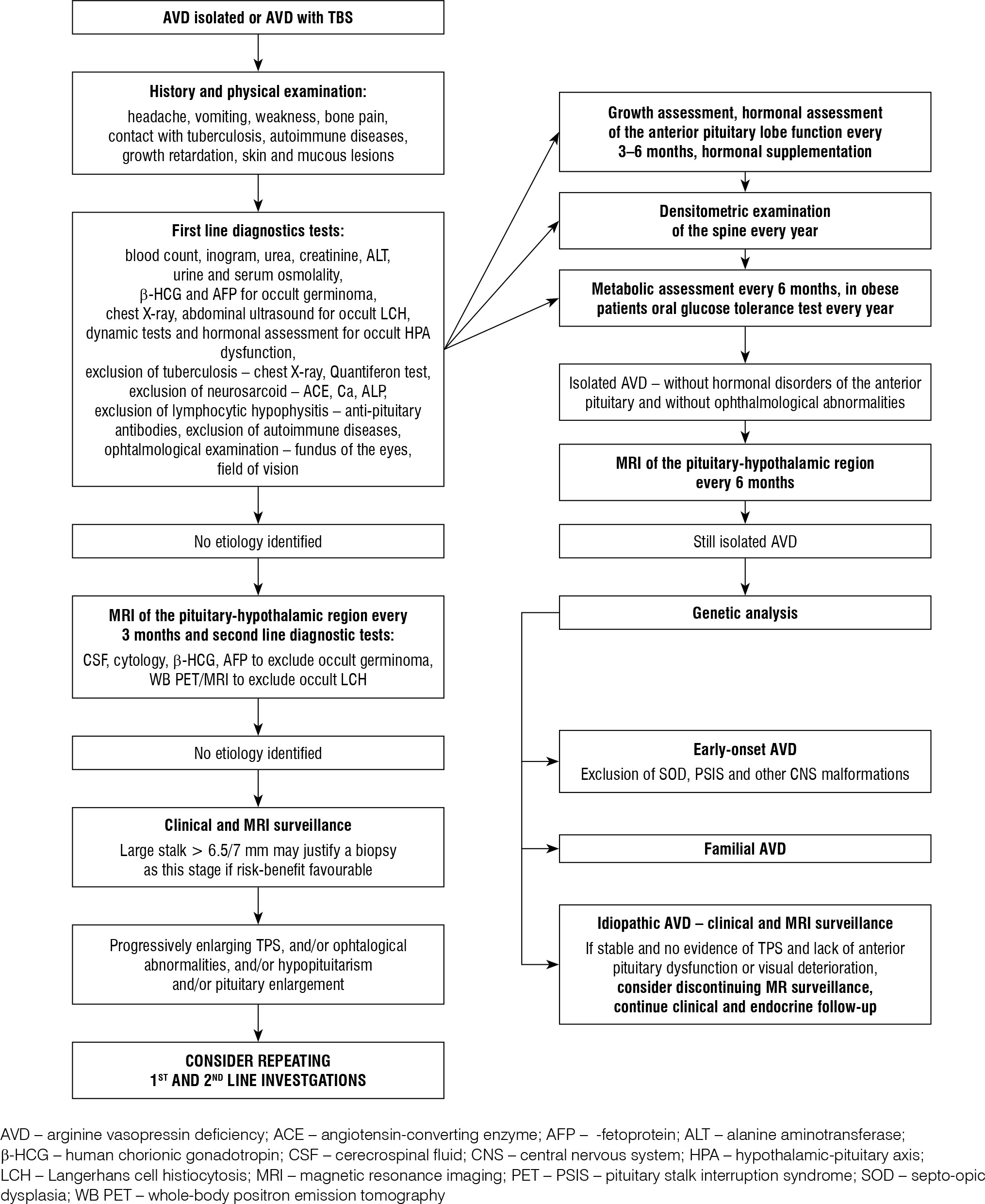
The presented report is based on a retrospective analysis of patients hospitalised in our centre from 1998 to 2023, and one of the main conclusions is that the time from the onset of symptoms to the diagnosis of AVD and the determination of the aetiology is too long. Our analysis, like other reports, indicates that germinoma and histiocytosis are the main causes of AVD in children [7–9]. To improve the initial diagnosis of AVD, cooperation between primary care physicians, endocrinologists, radiologists, oncologists, and neurosurgeons is necessary. In addition to a thorough interview indicating polydipsia and polyuria, attention should be paid to contact with patients with tuberculosis, the presence of autoimmune diseases in the family, and other accompanying symptoms: headaches, nausea, vomiting, slow growth, visual disturbances, drowsiness, and bone pain. In the physical examination, in addition to assessing the general condition, auxological analysis and neurological examination, hydration, and skin and mucosal changes should be assessed [2, 8] The diagnostic process involves the assessment of an ionogram, serum, and urine osmolality, a urine concentration test, and a vasopressin test, the accuracy of which is estimated at 70%; is not a perfect method [2]. In addition to the vasopressin dehydration test, an arginine test may be useful to determine copeptin (which is a stable C-terminal peptide of pre-pro vasopressin), and its concentrations correlate with AVP concentrations. In 2019, Winzeler et al. showed that a copeptin cut-off value of 3.8 pmol/l after arginine infusion had an accuracy of 93% in differentiating AVD from psychogenic polydipsia, with a sensitivity of 93% and a specificity of 92% [10]. Copeptin tests are not routinely available in all health care settings, and reference values have not yet been developed for children in specific age groups with normal weight, overweight, or obesity. However, there are studies that try to set reference ranges for children [11, 12].
Magnetic resonance imaging of the pituitary and brain is considered the gold standard in the diagnosis of AVD; the hyperintense signal of the posterior pituitary is explained by the storage of vasopressin in a complex with neurophysin II with a high lipid content. In 1994, a study was published that assessed the vascularisation of the pituitary gland in children with various conditions such as pituitary insufficiency, AVD, and LCH, compared to a control group. Dynamic magnetic resonance imaging was used to evaluate pituitary enhancement after administration of gadopentetate dimeglumine contrast. The results showed differences in pituitary enhancement times between patients with these conditions and the control group, suggesting abnormal vascularisation in the pituitary arteries and portal system [13].
In 9.6% of patients in our group, the absence of posterior lobe luminescence was the only finding associated with AVD. There are reports in the literature where, out of 138 patients with AVD, 12.2% had no cause for AVD and no high signal of the posterior pituitary lobe; and in other reports idiopathic AVD was diagnosed in 30% of patients [4, 14]. In the latest reports the percentage of diagnoses of idiopathic AVD is decreasing, which is due to more accurate diagnostics and the extension of the follow-up period in these patients [2].
Performing frequent check-ups in the first period after the diagnosis of idiopathic AVD plays a key role in the diagnosis of occult forms of germinomas and histiocytosis. In 2021 guidelines were published for children with pituitary stalk thickening and/or AVD, in which, in idiopathic AVD without TBS and without dysfunction of the anterior pituitary, a 3-year observation period is recommended, while in the case of thickening of the pedicle < 4.5–5 mm a 5-year period for observations is recommended [7]. In our 2 patients, the pituitary stalk thickening persisted for 5 years before the diagnosis of LCH (patient no. 5) and germinoma (patient no. 8). Other studies indicate the need for 10 years of follow-up of the pituitary stalk thickening, which may persist for up to 10 years in the case of histiocytosis and may indicate the need to look for changes outside the pituitary gland [2, 4]. Oncological guidelines have been prepared for patients with pituitary stalk thickening and/or AVD, in which the first line of diagnosis includes a blood analysis (ionogram, ESR, urea and creatinine levels), assessment of β-HCG and AFP levels in the blood serum, chest X-ray, abdominal ultrasound, ophthalmological examination (visus, fundus of the eyes, visual field), and bone scintigraphy. In the case of progressive pituitary stalk thickening > 6.5–7 mm and AVD, another diagnostic step is recommended, consisting of bone marrow puncture to assess cytosis, β-HCG, and AFP in the cerebrospinal fluid, and whole-body PET/MRI examination. Pituitary biopsy should be considered when the stalk thickening is > 7 mm and the benefits of biopsy outweigh the risk of CNS damage. In patients with idiopathic AVD, without extension of the pituitary stalk thickening and without other additional symptoms, follow-up NMR examinations of the pituitary and CNS should be repeated every 6 months, which will enable early diagnosis of the aetiology of AVD, earlier diagnosis of the tumour and prevent further damage to the CNS [2, 7]. It is worth extending the differential diagnosis of AVD by excluding tuberculosis and sarcoidosis by performing a Quantiferon test, chest X-ray, head CT, and checking the concentrations of Ca, ALKP, and ACE-converting enzyme activity [8]. For the diagnosis of germinomas, apart from the determination of β-HCG and AFP in serum and cerebrospinal fluid other markers such as placental alkaline phosphatase or OCT4 protein are recommended [15, 16].
Another problem revealed by our analysis was the lack of frequent hormonal control of anterior pituitary activity. Reports from the literature indicate that stalk thickening > 4.5 mm requires the exclusion of multihormonal hypopituitarism [17]. Our analysis showed that too many patients did not undergo stimulation tests to assess GH secretion due to the oncological complications. The 2023 consensus confirmed the safety of using rhGH in patients with CNS tumours and suggests the timing of initiation of rhGH therapy following completion of cancer therapy or treatment of an intracranial tumour depends on many factors and should be individualised; this period may be as early as 3 months in children with radiologically proven stable craniopharyngiomas who have significant growth failure and metabolic disturbances; for other types of tumours, it is advisable to wait at least one year the initiation of rhGH [18]. In patients with multihormonal hypopituitarism after extensive neurosurgical procedures, GH concentration in stimulation tests is usually < 3 ng/ml. In these patients with severe GH deficiency, the use of rhGH is necessary not only for growth improvement, but also for metabolic reasons. An additional and very important argument is the introduction of an rhGH treatment program in Poland for adult patients with hypopituitarism. For this reason, patients will be able to continue treatment as adults, which is of great importance to avoid metabolic and cardiovascular complications of severe GH deficiency and to improve the quality of life [19].
Auxological analysis should include not only the assessment of growth, but also weight gain. In patients with AVD and obesity, there is not only an increased risk of insulin resistance and type 2 diabetes; but there is also a serious risk of thromboembolic complications, which was the cause of death of our patient no. 20 with adipsic AVD and obesity of BMI 39 kg/m2. Hypothalamic obesity is a common complication of extensive neurosurgical procedures and leads to serious metabolic complications. Additionally, the absence of polydipsia and polyuria, key symptoms of AVD, may delay the diagnosis and lead to serious complications of a chronic hyperosmolar state and discontinuation of desmopressin therapy due to the absence of polyuria increasing the risk of deep vein thrombosis [20–22]. Postoperative immobilisation, obesity, infection, prolonged hospitalisation, blood thickening, and changes in coagulation that may be caused by inadequate hormonal treatment in the postoperative period (with high doses of glucocorticoids, sex steroids, and DDAVP replacement) may contribute to the pathogenesis of thrombosis. Therefore, thromboprophylactic treatment after pituitary surgery during episodes of hypernatraemia is suggested in adult patients with AVD [21, 22]. Assessment of the quality of life of adult patients after treatment of expansive processes initially manifesting as thickening of the pituitary stalk and/or AVD showed that the most common complication in this group was metabolic syndrome [8].
Few reports indicate a decrease in bone mineral density in patients with AVD. Decreased concentrations of osteocalcin (OC) and increased concentrations of cross-linked N-telopeptides of type I collagen (Ntx) have been demonstrated in the serum of patients with AVD compared to the control group, which indicates a reduction in bone formation in patients with AVD. Densitometric examination of the lumbar spine showed osteoporosis in 33.3% of patients and osteopaenia in 55.5% of patients with AVD. Another study described the positive effects of bisphosphonates in patients with AVD and low bone mass [20]. In our study, densitometric assessment was performed in children with AVD and anterior pituitary insufficiency. GH deficiency, hypogonadotropic hypogonadism, hydrocortisone supplementation in central adrenal insufficiency, and thyroxine in the treatment of secondary hypothyroidism are additional risk factors for osteoporosis. A pharmacoepidemiological study (the Pharmacia & Upjohn International Metabolic Database [KIMS]) assessing bone mineral density and the risk of fractures in 2024 adult patients with hypopituitarism showed that the risk of fractures in patients with GH deficiency is 2.66 times higher than in the control group [24]. Patients with AVD showed a significantly higher risk of fractures in men but not in women, but the overall percentage of patients with low BMD was similar between the AVD and non-AVD groups (14% vs. 13%). Although the mechanism behind this phenomenon is unknown, reduced bone density has been observed in patients with AVD [6]. Additional indications for densitometric examinations in patients with AVD are neurological deficits resulting from CNS damage; immobilisation, limb paresis, and visual disturbances limiting physical activity in children are further risk factors for low bone mass and fractures in adulthood [25].
Intact thirst protects self-sufficient patients from severe hypernatraemic dehydration, placing infants and patients with neurological disorders at greater risk of clinically significant dehydration [6]. Septo-optic dysplasia, considered part of the spectrum of congenital midline disorders, is the most common congenital malformation causing AVD, but only 10–15% of these patients present with symptoms of AVD. Much more often, in 60–80% of cases, there is hypofunction of the anterior pituitary lobe, which should be diagnosed and treated early [4]. Ectopia of the posterior lobe may also be a component of PSIS (pituitary stalk interruption syndrome), a rare entity that may be caused by mutations in many genes involved in the embryogenesis of the anterior lobe. The clinical picture of individual cases differs and changes over time, because the initial GH deficiency may be accompanied by symptoms of deficiency of other axes. Lack of proper diagnosis and appropriate treatment may pose a threat to the child’s life or cause permanent disability, as exemplified by the course of the disease and death of patient no. 21.
Genetic testing should be performed in patients with a positive family history and early-onset AVD [2]. So far, 80 gene mutations have been described that may result in vasopressin deficiency. AVP-NPII gene mutations are more often autosomal dominant (with the average onset of symptoms after the first year of life), less often autosomal recessive (presenting already between 1-2 weeks of age) or linked to the X chromosome. One of the causes may also be Wolfram syndrome, which results in atrophy of the hypothalamic nuclei, and degeneration of the optic nerves, pons, and cerebellum [2, 3].
There are reports about the importance of immunological diagnostics, in which antibodies against cells involved in the production and secretion of ADH were identified in 75% of paediatric patients with idiopathic central diabetes. However, the same antibodies were found in patients with established etiologist such as Langerhans cell histiocytosis, germinoma, or even in postoperative hypothalamus cases.
Conclusions
Based on 25 years of analysis we conclude the following:
Establishing the aetiology of AVD is difficult and often results in late diagnosis.
Rapid diagnosis of AVD is crucial in cases of oncological causes of AVD. MRI scans and available diagnostic schemes are the basis for efficient diagnosis.
Periodic assessment of the function of all axes of the hypothalamus-pituitary axis is recommended, along with the prompt initiation of hormonal substitution.
Metabolic control and obesity prevention are necessary for children with AVD.
For patients with AVD, densitometric monitoring and osteoporosis prevention should be recommended.
Therefore, based on the above-mentioned conclusions and using the presented diagnostic diagrams [8, 9], we have developed a scheme for hormonal and metabolic diagnostics of patients with AVD, which may prove helpful not only for earlier determination of the aetiology of AVD, but above all should serve to prevent hormonal, metabolic, and bone complications in this group of patients, which is the basis for the comfort of life in developmental age and adulthood (Fig. 3).