
Introduction
Myelodysplastic neoplasms (MDS) comprise haematological neoplasms characterised by morphologic dysplasia, cytopaenia(s), hypercellularity, and the risk of transformation into acute myeloid leukaemia myelodysplasia- related (AML-MR) [1]. Accurate risk-prognosis tools [2–4] divide patients into lower-risk myelodysplastic neoplasms (LR-MDS) (two-thirds of patients) and higher-risk MDS (HR-MDS), associated with entirely different treatment approaches [5]. In LR-MDS, the treatment strategy aims to alleviate cytopaenia, reduce the need for blood product support, and improve patients’ life quality. Despite the generally favourable prognosis of LR-MDS, some patients are affected by relatively fast progression [6].
At least one pathogenic mutation is observed in 64% of LR-MDS patients, with the total number of mutations ranging 0–9 [7]. SF3B1 mutation represents a marker of favourable prognosis in LR-MDS. However, co-mutations in RUNX1 or EZH2 have been associated with rapid progression [6–8]. Moreover, ASXL1, TP53, SRSF2, and RUNX1 mutations are independently associated with poor prognosis [9]. In addition, multiple mutations present in a single LR-MDS patient further predispose to poor outcome [10].
The presented study was designed to determine the mutational profile of LR-MDS patients using next-generation sequencing (NGS), pyrosequen- cing, and Sanger Sequencing (SSeq). We aimed to identify clinical implications of detected variants in the LR-MDS diagnosis and progression. We have discussed the usefulness of peripheral blood (PB) and saliva (SAL) samples for genetic reassessment of LR-MDS patients.
Material and methods
Study group
The study was approved by the local Bioethical Committee (approval no. 536/14), in accordance with the Declaration of Helsinki. All participants provided informed written consent.
The study group comprised 30 primary MDS patients diagnosed in centres within the Polish Adult Leukaemia Group. The inclusion criteria were as follows: MDS diagnosis (according to 2016WHO criteria), age ≥ 18 years, hospitalisation between 09.2014 and 09.2021, and a bone marrow (BM) sample collected for genetic testing. Lower-risk myelodysplastic neoplasms were defined as IPSS-R score ≤ 3.5.
Clinical data included: age, gender, diagnosis, molecular/ cytogenetic data, treatment type and response, transplant-related features, and MDS progression according to the revised International Working Group criteria [11, 12], including progression to AML (2016 WHO criteria). The analysis of overall survival (OS) was also conducted. We also implemented 2022 WHO criteria for patient diagnosis [1].
Sample collection
The study group comprised 23 LR-MDS and 7 HR-MDS. At the moment of analysis, the samples included 22 LR-MDS, 5 HR-MDS, and 3 AML-MR (one prior LR-MDS and 2 prior HR-MDS). Material collected from 23 LR-MDS patients comprised 25 BM, 3 SAL, and one PB. Bone marrow of 2 patients (MDS-9A, B and MDS-13A, B) was collected twice, at different disease stages. DNA samples isolated from the PB of 5 healthy controls were used to establish the limit of allele frequency (AF) detection using pyrosequencing.
DNA isolation
DNA was isolated based on a phenol-chloroform mixture and isopropanol precipitation procedure and stored at –20°C.
Next-generation sequencing
Nine genes most frequently mutated in MDS (Table 1) were selected to perform targeted NGS. Sixty-seven exons (Table 1) were sequenced (pair-end) using targeted NGS (MiSeq system (Illumina), sequence coverage: 31-9095, reading length: 250 bp) in 12 patients: 4 LR-MDS, 5 HR-MDS, and 3 AML-MR (2 prior HR-MDS and one prior LR-MDS). Bioinformatic analyses comprised read mapping to reference genome (hg19), using the bwa-mem algorithm (BWA,0.7.10) [https://pubmed.ncbi.nlm.nih.gov/20080505/]. Detection of variants was performed using GATK software (v.3.5) [https://pubmed.ncbi.nlm.nih.gov/25431634/] with further annotation using snpEff (4.2) [https://pubmed.ncbi.nlm.nih.gov/22728672/], based on the dbSNP142 database. For further analysis, only nonsynonymous substitutions and indels, novel variants, or those already annotated with mean AF < 1% were selected.
Table 1
Regions analysed by pyrosequencing
Confirmation of next-generation sequencing-detected DNA sequence variants using Sanger Sequencing
Sanger Sequencing was performed to confirm the presence of DNA sequence variants (DNA-seq-var) detected by NGS. Primers for amplicon preparation were designed using the Primer3Plus tool (https://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi).
Sanger Sequencing analyses were performed according to the described protocol [13]. Sequencing results were analysed with the use of CodonCodeAligner software with the reference sequence.
Pyrosequencing and Sanger Sequencing testing for the presence of DNA sequence variants in an extended cohort
To verify whether detected DNA-seq-var could be classified as hot-spots for LR-MDS/AML-MR, we performed SSeq and pyrosequencing genotyping within all 23 primary LR-MDS patients. The Sanger Sequencing was performed as described above. PyroMark Assay Design Software 2.0.1.15 (Qiagen) was used to design primers for pyrosequencing. The primer set was composed of primers for amplicon preparation (including one labelled with biotin at the 5’end) and sequencing primer (Table 1). The pyrosequencing tests were designed using the PyroMarkQ48 Autoprep 2.4.2 Software (Qiagen). Pyrosequencing was performed using PyroMarkQ48 Advanced CpG Reagents (Qiagen) and PyroMarkQ48 Autoprep (Qiagen), according to the manufacturer’s standard protocol, with quantified mean mutated AF determined. Sample positivity thresholds were calculated for each pyrosequencing test, based on healthy control results (5 PB). The allele frequency threshold was calculated as twice the standard deviation plus the highest AF value obtained for healthy controls. DNA sequence variants previously detected in BM were additionally genotyped in one matched PB and 3 SAL samples.
Results
Characteristics of lower-risk myelodysplastic neoplasms
Median age at LR-MDS diagnosis was 65.6 years. Acute myeloid leukaemia myelodysplasia-related progression occurred in 3/23 cases, after median time of 31 months (Table 2). Cytogenetic results are presented in Table 3.
Table 2
Clinical characteristics of myelodysplastic neoplasms
[i] A – androgens, alloHCT – allogenic hematopoietic cell transplantation, AZA – azacitidine, CSS MDS – comprehensive cytogenetic scoring system for myelodysplastic neoplasms, CTH – chemotherapy, DS-SLD – myelodysplastic neoplasms with single lineage dysplasia, ESA – erythroid stimulating agents, G – good, h-MDS – hypoplastic myelodysplastic neoplasms, I – intermediate, L – luspatercept, LEN – lenalidomide, MDS-EB1 – myelodysplastic neoplasms with excess of blasts 1, MDS-EB2 – myelodysplastic neoplasms with excess of blasts 2, MDS-h – hypoplastic myelodysplastic neoplasms, MDS-IB1 – myelodysplastic neoplasms with increased blasts 1, MDS-LB – myelodysplastic neoplasms with low blasts, MDS-MLD – myelodysplastic neoplasms with multilineage dysplasia, MDS-RS-MLD – myelodysplastic neoplasms with ring sideroblasts and multilineage dysplasia, MDS-RS-SLD – myelodysplastic neoplasms with ring sideroblasts and single lineage dysplasia, MDS-SF3B1 – myelodysplastic neoplasms with SF3B1 mutation, MDS-U – myelodysplastic neoplasms unclassified, MP – poor, T-AML-MR – acute myeloid leukemia myelodysplasia-related transformation, VG – very good, VP – very poor
Table 3
Cytogenetic characteristics of lower-risk myelodysplastic neoplasms
Treatment implementation
For LR-MDS treatment, erythroid stimulating agents, lenalidomide or luspatercept, were used. After disease progression, treatment with hypomethylating agents and intensive treatment was implemented. Three patients underwent allogenic hematopoietic cell transplantation (alloHCT) (Tables 2, 4).
Table 4
Allogenic hematopoietic cell transplantations in lower-risk myelodysplastic neoplasms
[i] allo-haploHCT – haploidentical allogenic hematopoietic cell transplantation, alloHCT – allogenic hematopoietic cell transplantation, CSS MDS – comprehensive cytogenetic scoring system for myelodysplastic neoplasms, h-MDS – hypoplastic myelodysplastic neoplasms, LR-MDS – lower-risk myelodysplastic neoplasms, MAC – myeloablative conditioning, MDS-RS-MLD – myelodysplastic neoplasms with ring sideroblasts and multilineage dysplasia, RIC – reduced-intensity conditioning
Next-generation sequencing results
At least one genetic alteration was detected in 55.6% of MDS and 66.6% of AML-MR patients. The number of detected DNA-seq-var (BM) ranged 0–4. Altogether, 13 DNA-seq-var were detected in the 7 genes (Table 5).
Table 5
Next generation sequencing results
In the LR-MDS subgroup, at least one genetic alteration was found in 75.0% of MDS patients and one AML-MR (after LR-MDS). The number of detected DNA-seq-var (BM) ranged 0–4. Eight DNA-seq-var were detected in 6 genes (Table 5).
According to the COSMIC database, 7 DNA-seq-var were not previously described in MDS: c.1014+1G>T DNMT3A, c.509-2A>C RUNX1, c.1945G>T ASXL1, c.2757dupA ASXL1, c.4638G>C TET2, c.4044+2dupT TET2, and c.4076G>T TET2. c.2390A>G. While DNMT3A was previously reported in AML-MR, and c.1945G>T ASXL1 was detected in AML [14], in our study they were both present in LR-MDS. Similarly, c.509-2A>C RUNX1 was previously described in AML, whereas we detected it in both AML-MR and LR-MDS [15]. All DNA-seq-var detected using NGS were confirmed using SSeq and pyrosequencing.
Pyrosequencing and Sanger Sequencing testing in a larger cohort
Lower-risk myelodysplastic neoplasm analysis revealed the presence of 5 DNA-seq-var at the MDS disease stage. Within the LR-MDS subgroup, one DNA-seq-var was detected in 3 patients, and 2 DNA-seq-var were detected in one patient. Five DNA-seq-var were found in LR-MDS case that progressed to AML-MR (4 DNA-seq-var were detected initially, with acquisition of a fifth DNA-seq-var observed later) (Table 6) [15].
Table 6
Positive results of genotyping of lower-risk myelodysplastic neoplasms and acute myeloid leukemia myelodysplasia-related patients with pyrosequencing and Sanger Sequencing
[i] AF – allele frequency, AML-MR – acute myeloid leukemia myelodysplasia-related, BM – bone marrow, HC – healthy controls, LR-MDS – lower-risk myelodysplastic neoplasms, MUT – mutated, NGS – mutation detected by next generation sequencing in bone marrow, P – pyrosequencing, PB – peripheral blood, SAL – salive, Sseq – Sanger Sequencing, WT – wild-type; * Pyrosequencing results defined as AF (%).
Detailed clinical characteristics of patients, accompanied by positive genetic results, are listed in Table 7.
Table 7
Clinical characteristics and cytogenetic results of patients with positive genomic results
| Category | Sample | NGS (mutations n) | MDS subtype | Cytogenetics | AML-MR transformation (months) | Median OS (months) | Treatment |
|---|---|---|---|---|---|---|---|
| MDS | MDS-9A,B | Yes (2) | MDS-5q- | del5q | Yes (34) | 35 (death) | CTH, AZA, LEN |
| MDS-29 | Yes (1) | MDS-MLD | unanalysable | No | 103 (alive) | n/a | |
| MDS-30 | Yes (1) | MDS-MLD | unanalysable | No | 147 (alive) | ESA | |
| MDS-102 | No | MDS-RS-SLD | 46,XY[19] | No | n/a | n/a | |
| AML-MRC | MDS-13A,B | Yes (4) | MDS-RS-MLD/ MDS-EB2 | 46,XY[10] | Yes (31) | 123 (death) | CTH, alloHCT, AZA |
[i] alloHCT – allogenic he matopoietic cell transplantation, AML-MR – acute myeloid leukemia myelodysplasia-related, AZA – azacitidine, CTH – chemotherapy, ESA – erythroid stimulating agents, LEN – lenalidomide, MDS-EB2 – myelodysplastic neoplasms with excess of blasts 2, MDS-MLD – myelodysplastic neoplasms with multilineage dysplasia, MDS-RS-MLD – myelodysplastic neoplasms with ring sideroblasts and multilineage dysplasia, MDS-RS-SLD – myelodysplastic neoplasms with ring sideroblasts and single lineage dysplasia, n/a – no data, NGS – next generation sequencing, OS – overall survival
Peripheral blood and saliva results
One DNA-seq-var, detected previously in BM, was confirmed using SSeq in 1) PB (one out of one case, sample MDS-9A), and 2) SAL (2 out of 2 cases, samples MDS-9A, MDS-13A). Pyrosequencing confirmed the presence of all DNA-seq-var detected previously in BM in sample MDS-13A, and only one DNA-seq-var in sample MDS-13B [15]. Allele frequency levels of mutations detected using pyrosequencing were slightly lower in SAL than in BM (Table 6).
Survival of lower-risk myelodysplastic neoplasms
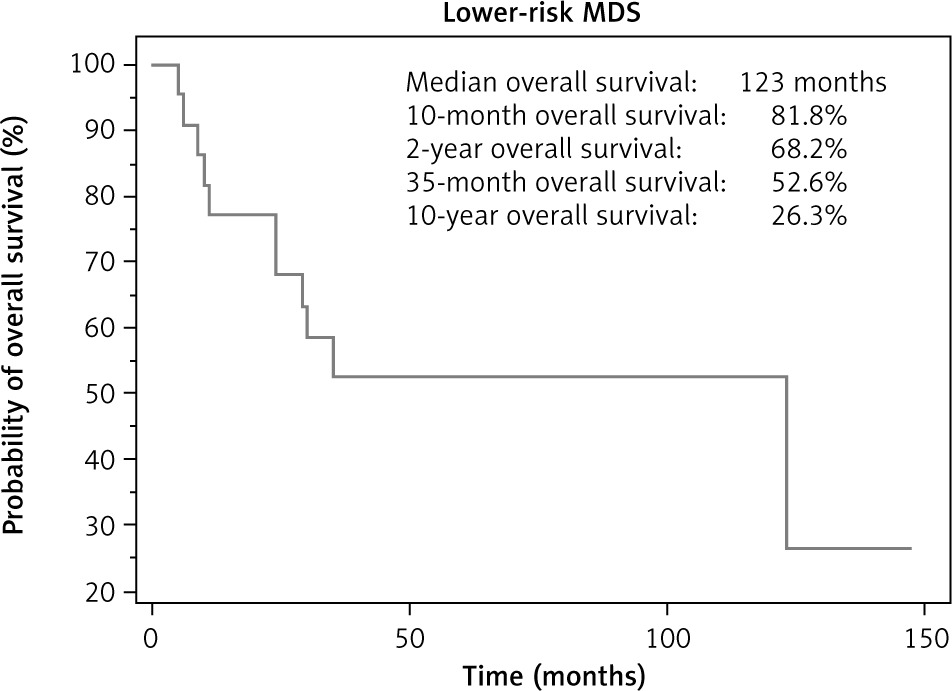
The median follow-up was 24 months, and the median OS for LR-MDS patients was 123 months (Fig. 1).
ELN2017 and ELN2022 genetic risk stratification for acute myeloid leukaemia myelodysplasia-related
Lower-risk myelodysplastic neoplasms patients with AML-MR progression were classified as intermediate (n = 1) or adverse (n = 2) subgroups, according to both the ELN2017 and the ELN2022. Considering the results of our genomic study, all AML-MR patients were reclassified to the adverse subgroup (n = 3) due to the co-occurrence of ASXL1, RUNX1, and SF3B1 DNA-seq-var (Table 6).
Discussion
Our study was based on a clinical analysis of patients with LR-MDS, tracking its course, including AML-MR progression, in relation to genetic findings. We examined NGS clinical testing utility in LR-MDS. We evaluated PB and SAL testing as useful methods for serial reassessment of LR-MDS patients at risk of disease progression.
The median number of observed mutations within LR-MDS patients usually ranges 0–9 [7, 16], while only 1–2 DNA-seq-var were detected in our study.
It was previously reported that the presence of RUNX1 and TP53 mutations contributes to LR-MDS progression to AML-MR [17]. We noted the acquisition of RUNX1 c.509-2A>C mutation during the course of LR-MDS (patient MDS-13) [15]. Lower-risk myelodysplastic neoplasm patients with the RUNX1 mutation detected at diagnosis should be subject to intensive and strict monitoring [7].
ASXL1 and RUNX1 mutations play important roles in the development of AML-MR [18]. Both these mutations were detected in LR-MDS, and after disease progression to AML-MR. The SF3B1 gene is one of most commonly mutated genes in LR-MDS, showing a more frequent mutation rate in MDS than in AML-MR in previous studies [7, 18, 19]. SF3B1 mutation was found in LR-MDS with ring sideroblasts (one MDS-RS-SLD and one MDS-RS-MLD/AML-MR patient) [15].
Cytogenetic assessment is an insufficient diagnostic tool in MDS because molecular biology methods comprise a significantly more precise differentiating tool. The recently introduced IPSS-M risk stratification in MDS incorporates mutational profiles of 31 genes, resulting in re-stratification of prognostic category in 46% of MDS patients (compared to IPSS-R) [4]. Performing genetic analysis at LR-MDS/AML-MR diagnosis and incorporating molecular findings into stratification tools remains crucial to predict patient outcomes. In our study, NGS testing revealed the presence of ZRSR2 mutation at diagnosis in one patient (MDS-29), with cytogenetic karyotyping impossible at this stage. Cytogenetic abnormalities (CA) occur in about 50% of cases [20], and in our study the CA frequency was 55.6%. Acquisition of CA in LR-MDS is associated with higher risk of AML-MR transformation and poor survival [21].
Lower-risk myelodysplastic neoplasms are characterised by long OS [22, 23], and our study reaffirmed this observation. Acute myeloid leukaemia myelodysplasia-related is characterised by a higher frequency of TP53 mutation than MDS [18], and high frequency of complex karyotype [24]. Our results indicate co-occurrence of complex karyotype after LR-MDS progression. We noted MDS-5q progression to AML-MR in 10% of MDS-5q patients [25].
Considering our molecular testing results, the adverse, instead of intermediate, classification was implemented according to both the ELN2017 and ELN2022. Even though LR-MDS are characterised by favourable prognosis, some patients can progress rapidly [6]. Clinical vigilance is required, and detection of adverse molecular markers at LR-MDS diagnosis should prompt consideration of serial reassessment [6]. Contrary to routinely performed BM aspiration, both PB and SAL can be noninvasively collected in LR-MDS patients who require careful monitoring. All DNA-seq-var, reported using NGS in BM, were also detected using SSeq and pyrosequencing in both BM and PB. Pyrosequencing results in patient MDS-13B in BM revealed low AF of 5/5 detected mutations; however, SAL analysis confirmed only 1/5 mutations. For pyrosequencing, AF values of the detected variants in BM and SAL were slightly lower for SAL but were nonetheless comparable. Saliva testing is limited by a low count of leukemic cells and, within alloHCT recipients, by the presence of high donor chimerism. Overall, the above-mentioned results suggest diagnostic utility of SAL and PB testing for monitoring LR-MDS cases at higher risk of disease progression. Hence, studies on a larger group are required.
Allogenic hematopoietic cell transplantation were performed in 3 primarily LR-MDS cases. In LR-MDS patients, alloHCT should be considered in the presence of BM fibrosis, adverse molecular features, increased red blood cell transfusion dependence, therapy-related disease, and after disease progression [21, 26, 27]. For LR-MDS, the genomic features may support decision-making because they allow the prediction of survival rates after alloHCT [28].
Limitations: The clinical data were heterogenous, the study group comprised only a narrow part of a larger genomic MDS study, and due to restricted funds, only 12 patients were tested using NGS, and only small number of genes was sequenced.
Conclusions
Our study defines the genetic findings in patients with LR-MDS and disease progression, in relation to the colle- cted clinical data. In cases of LR-MDS progression suspicion, we suggest using targeted NGS. Pyrosequencing enables accurate tracking of genetic alterations, previously detected by NGS, throughout the course of the disease. We also indicate the clinical utility of PB and SAL for genomic testing, as an alternative to BM for LR-MDS patients at higher risk of rapid disease progression. However, BM still appears to be the best diagnostic choice for MDS patients, allowing false-negative results to be avoided when the mutation occurs in small variant allele frequency. Finally, our study confirms the presence of mutational acquisition throughout the progression of LR-MDS into AML-MR.






