
Introduction
Hepatic encephalopathy (HE) is a severe clinical syndrome associated with a high mortality rate if not appropriately managed [1, 2]. Ammonia (NH4+) is the primary culprit responsible for organ injury during HE [1-3]. The brain is the principal organ affected by NH4+ [1-3]. Hyperammonemia could lead to severe central nervous system (CNS) complications ranging from behavioral and cognitive impairment to coma and death [3]. Although several strategies have been developed for decreasing blood NH4+ during HE, no specific pharmacological option is available to manage HE-associated brain injury. Therefore, searching for novel therapeutic options in this field is of importance.
The mechanism of NH4+-induced brain injury has been widely investigated [3-8]. It has been found that NH4+ could induce severe oxidative stress in the brain tissue [4, 9]. A significant amount of reactive oxygen species (ROS) formation, lipid peroxidation, and a decreased level of tissue antioxidants have been documented in experimental models of hyperammonemia [9-11]. Moreover, NH4+ could induce significant mitochondrial impairment in the brain [12-15]. The effects of NH4+ on neuroinflammation have also been documented in several HE models [3, 5, 12, 14, 16-19]. Mitochondrial impairment and neuroinflammation are also mechanistically interrelated with oxidative stress [3].
Dextromethorphan (DXM) is a CNS active agent widely used as an effective and safe cough suppressant [20]. Interestingly, several studies have revealed that DXM provides significant CNS antioxidant effects [21-26]. Various investigations have confirmed the neuroprotective properties of DXM in complications such as cerebral ischemia, Parkinson’s disease, multiple sclerosis, spinal cord injury, and epilepsy [21, 27-31]. It has been found that DXM significantly blunted ROS formation and lipid peroxidation in experimental models of CNS disorders [21, 27-31]. DXM also caused the upregulation of cellular antioxidant defense mechanisms (e.g., superoxide dismutase enzyme) [21, 27-31]. The effect of DXM on nuclear factor-erythroid 2 related factor 2 (Nrf2) as the primary mechanism responsible for the regulation of the cellular antioxidant defense mechanism seems to play a pivotal role in its antioxidant action [21, 27-31]. Therefore, the effects of DXM on oxidative stress biomarkers in the brain tissue of an animal model of HE are evaluated in the current study.
Previous studies also mentioned the effects of DXM in mitigating neuroinflammation in various investigations [32, 33]. It has been found that DXM significantly decreased brain levels of pro-inflammatory cytokines such as interleukin 1β (IL-1β), tumor necrosis factor α (TNF-α), and interleukin 6 (IL-6) [32]. In the current study, the effect of DXM on brain pro-inflammatory cytokines is investigated to shed light on the potential mechanism of action of this drug in the brain of hyperammonemic animals.
As NH4+-induced oxidative stress and neuroinflammation play a crucial role in the pathogenesis of HE, the current study was designed to evaluate the effects of DXM in the management of locomotor dysfunction, brain oxidative stress biomarkers, and inflammatory cytokines in an animal model of acute liver failure and hyperammonemia. The results could help the development of pharmacological interventions in HE.
Material and methods
Chemicals
4,2-Hydroxyethyl,1-piperazine ethane sulfonic acid (HEPES), thiobarbituric acid, 6-hydroxy-2,5,7,8-tetra- methyl chroman-2-carboxylic acid (Trolox), dithiobis-2-nitrobenzoic acid (DTNB), malondialdehyde, glutathione (GSH), 2’,7’-dichlorofluorescein diacetate (DCFH-DA), potassium chloride (KCl), sodium phosphate dibasic (Na2HPO4), sodium phosphate monobasic (NaH2PO4), dextromethorphan hydrobromide monohydrate, and ethylenediaminetetraacetic acid (EDTA) were purchased from Sigma (St. Louis, MO, USA). Trichloroacetic acid and hydroxymethyl aminomethane hydrochloride (Tris-HCl) were purchased from Merck (Darmstadt, Germany). Commercial kits were used to assess plasma biomarkers (Pars Azmoon, Tehran, Iran) and brain levels of cytokines (Shanghai Jianglai Biology, China).
Animals
Mature male BALB/c mice (n = 36, weighing 25 ±3 g) were obtained from Shiraz University of Medical Sciences, Shiraz, Iran. Animals were maintained in a standard environment (12 : 12 light : dark cycle, temperature 23 ±1°C, and 40 ±5% relative humidity) with free access to tap water and a standard diet (Behparvar, Tehran, Iran). All animal procedures were conducted under the supervision of the institutional ethics committee of Shiraz University of Medical Sciences, Shiraz, Iran (IR.SUMS.REC.1399.1352).
Experimental setup
A single dose of acetaminophen (APAP; 1000 mg/kg, IP) was used to induce acute liver failure [34]. DXM (0.5, 1, 5, and 10 mg/kg, subcutaneously [SC], three doses with 6 h intervals) [26], was administered one hour after acetaminophen. Animals’ locomotor activity was evaluated 24 hours after APAP treatment. Then, animals were anesthetized (thiopental, 80 mg/kg, intraperitoneally [IP]), and blood and brain samples were collected for further analysis.
Animals’ locomotor activity
Rotarod test
Based on a previously reported protocol, animals underwent three sessions of the rotarod test to evaluate their locomotor function in HE [35, 36]. The speed of the rotating rod was 5 and 10 rpm with a cut-off point of 300 s [35, 36]. Each test period for each animal consisted of three episodes with 10 min intervals. The time that each mouse stayed on the rotating rod was automatically recorded [35, 36].
Open-field behavior test
The open-field behavior test was used as an index of animals’ locomotor activity in the current model of hyperammonemia and HE [37]. The open field apparatus was made of a white box (1 m L × 1 m W × 0.3 m H). The box floor was divided into 25 squares (25 × 25 cm), and the open field was equipped with a webcam (2.0 Megapixel, Gigaware, UK). Animals’ activities were monitored and recorded for 15 minutes. The number of crossed squares was recorded as total locomotion in the mentioned time frame [37, 38].
Gait stride test
Gait stride was measured as an index of locomotor function in HE [36, 37]. For this purpose, mice’s hind paws were wetted with ink, and animals were allowed to walk down on a paper strip (60 cm long, 10 cm wide) from the brightly lit corridor toward the dark side. The distances between the points of the left and right hind paws were recorded [36, 39].
Sample collection
Animals were deeply anesthetized with thiopental (80 mg/kg, IP). The blood sample was collected from the abdominal vena cava. Blood was transferred to heparin-coated tubes and centrifuged (4000 g, 10 min, 4°C). The plasma was used for biochemical analyses. The brain tissue was isolated and washed with ice-cooled normal saline solution (0.9% NaCl, 4°C) and used for further analysis.
Biochemical analysis
Commercial kits (Parsazmoon, Tehran, Iran) and a MindrayBS-200 autoanalyzer were used to measure plasma aspartate aminotransferase (AST), alanine aminotransferase (ALT), and bilirubin. The plasma ammonia level was measured based on the method of phenate-hypochlorite reaction [37, 40]. For the determination of brain ammonia content, samples (100 mg) of the forebrain (cerebral cortex) were homogenized and deproteinized in 3 ml of ice-cooled lysis solution (trichloroacetic acid, 6%, w/v, 4°C). After centrifugation (16,000 g, 10 minutes, 4°C), the supernatant was collected, neutralized with potassium carbonate (100 µl of KHCO3; 2 mol/l, pH = 7), and used for assessing ammonia levels [37]. Brain tissue levels of TNF-α, IL-6, and IL-1β were measured using commercial kits (Shanghai Jianglai Biology, China) based on the manufacturer’s instructions.
Brain tissue lipid peroxidation
Thiobarbituric acid reactive substances (TBARS) were measured as an index of lipid peroxidation in the brain tissue [41-43]. Briefly, 250 mg of the brain tissue was homogenized in 2.5 ml of TrisHCl buffer (40 mM, pH = 7.4). Afterward, 500 µl of tissue homogenate was treated with thiobarbituric acid (500 µl of 0.6%, w : v), and 500 µl of meta-phosphoric acid (1% w : v, pH = 2) [44-46]. Samples were mixed well and heated in a water bath (100°C; 45 min). Then, the mixture was cooled, and 500 µl of n-butanol was added. Samples were vigorously vortexed and centrifuged (16000 g, 5 min). Finally, the absorbance of the upper phase was measured at λ = 532 nm (EPOCH plate reader, US) [47].
Reactive oxygen species (ROS) formation in the brain tissue
2’,7’-dichlorofluorescein-diacetate (DCF-DA) as used as a fluorescent probe to estimate brain ROS levels [48-51]. Brain tissue was homogenized in 2.5 ml of ice-cooled (4°C) Tris-HCl buffer (40 mM, pH = 7.4).Samples (100 µl) of the resulting tissue homogenate were mixed with Tris-HCl buffer (1 ml; pH = 7.4, 4°C) and 5 µl of DCF-DA (final concentration of 10 µM) [52-56]. The mixture was incubated at 37°C (15 min, in the dark). Finally, the fluorescence intensity was measured using a FLUOstar Omega plate reader (λ excitation = 485 nm and λ emission = 525 nm) [57, 58].
Brain tissue glutathione (GSH) content
Brain tissue GSH content was assessed using Ellman’s reagent (dithiobis-2-nitrobenzoic acid, DTNB) [59-61]. Briefly, 500 µl of the prepared brain homogenate was treated with 100 µl of trichloroacetic acid (50% w : v; 4°C). Samples were mixed well and centrifuged (6000 g, 4°C, 15 min). Afterward, the supernatant was mixed with 4 ml of Tris-HCl buffer (pH = 8.9; 4°C) and 100 µl of DTNB (dissolved in methanol). Samples were mixed well, and the absorbance was measured at λ = 412 nm (EPOCH plate reader, US) [62].
Brain tissue antioxidant capacity
The total antioxidant capacity of the brain tissue was assessed using the ferric reducing antioxidant power (FRAP) test [63-66]. Briefly, 100 µl of the tissue homogenate was added to 1000 µl of FRAP reagent (composed of 250 µl of acetate buffer 300 mM, 250 µl of iron chloride solution, and 250 µl of 10 mM TPTZ in 40 mM HCl). The mixture was incubated at 37°C (5 min, in the dark). Finally, samples were centrifuged (16000 g, 1 min), and the absorbance of the developed color was measured at λ = 593 nm (EPOCH plate reader, US) [63, 64].
Results
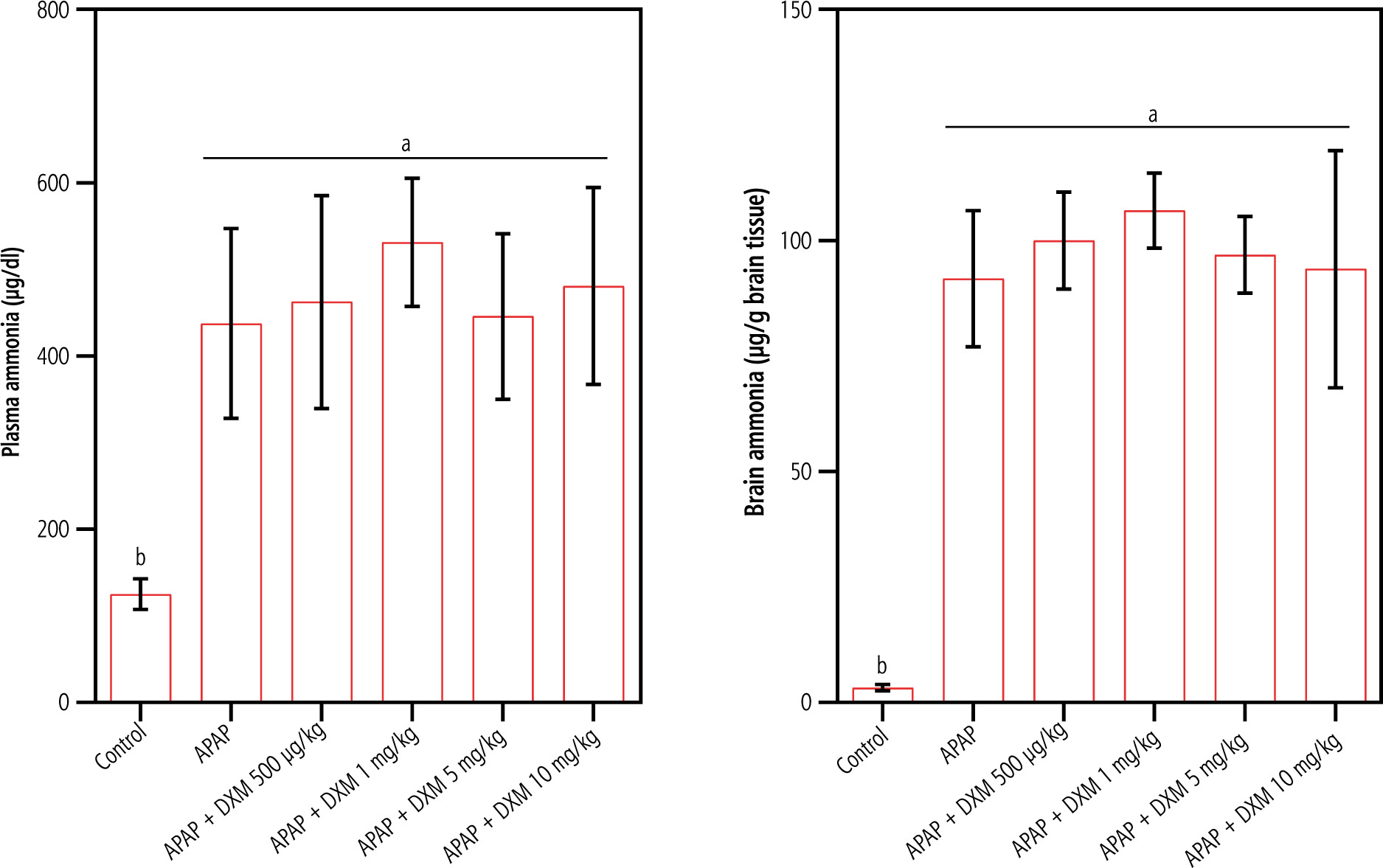
Significant changes in serum biomarkers of liver injury, including increased ALT, AST, and bilirubin, revealed marked hepatic damage in APAP-treated animals (Table 1). It was found that DXM (0.5, 1, 5, and 10 mg/kg, SC) caused no significant changes in serum biomarkers of liver injury in APAP-treated animals (Table 1). On the other hand, a significant increase in plasma and brain ammonia levels was detected in the APAP-treated mice (Fig. 1). DXM (0.5, 1, 5, and 10 mg/kg, SC) caused no significant change in plasma and brain ammonia levels (Fig. 1).
Table 1
Serum biochemistry in acetaminophen (APAP)-treated mice
Fig. 1
Acetaminophen (APAP; 1 g/kg, IP) caused a significant increase in plasma and brain tissue ammonia levels in mice. In the current model, dextromethorphan (DXM) had no significant effects on blood and brain ammonia. Data are expressed as mean ±SD (n = 6). Different alphabetical superscripts indicate that the mentioned groups are significantly different (p < 0.05)
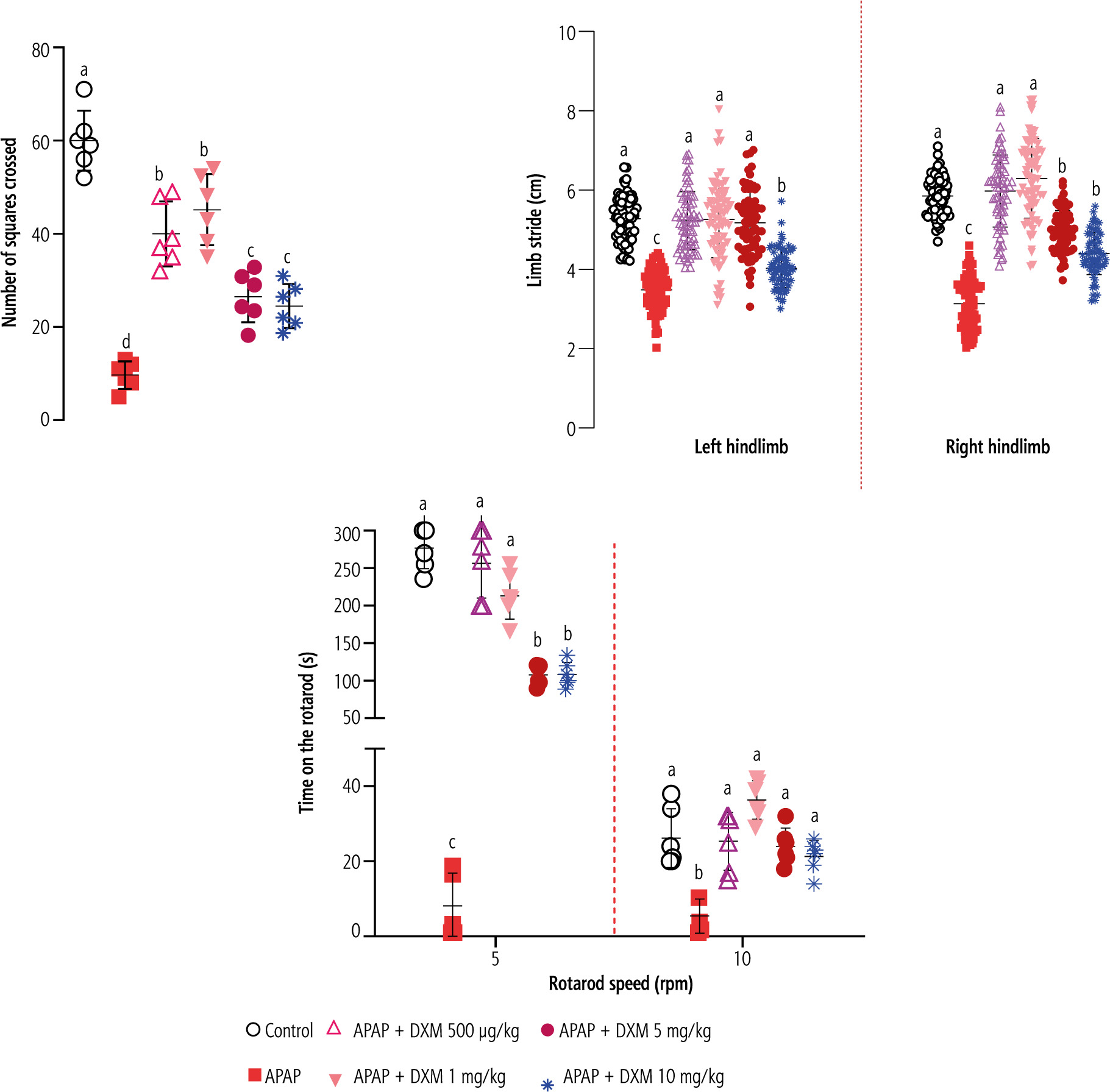
Evaluating animals’ locomotor activity in APAP-treated mice revealed a significant decrease in open-field behavior, the distance of the left and right hind paw marks (gait stride test), and the time that animals stayed on the rotating rod. It was found that DXM (0.5, 1, 5, and 10 mg/kg) significantly improved animals’ locomotor activities. The effects of DXM on animals’ locomotor activity were more prominent in lower doses of this drug (0.5 and 1 mg/kg) (Fig. 2).
Fig. 2
Locomotor activity in acetaminophen (APAP)-treated mice. A significant decrease was detected in the animals’ locomotor activity in APAP-treated animals. It was found that dextromethorphan (DXM) significantly improved animals’ locomotion in APAP-treated groups. Data are expressed as mean ±SD (n = 6). Data for limb stride are shown for at least n = 70. Different alphabetical superscripts indicate that the mentioned groups are significantly different (p < 0.05)
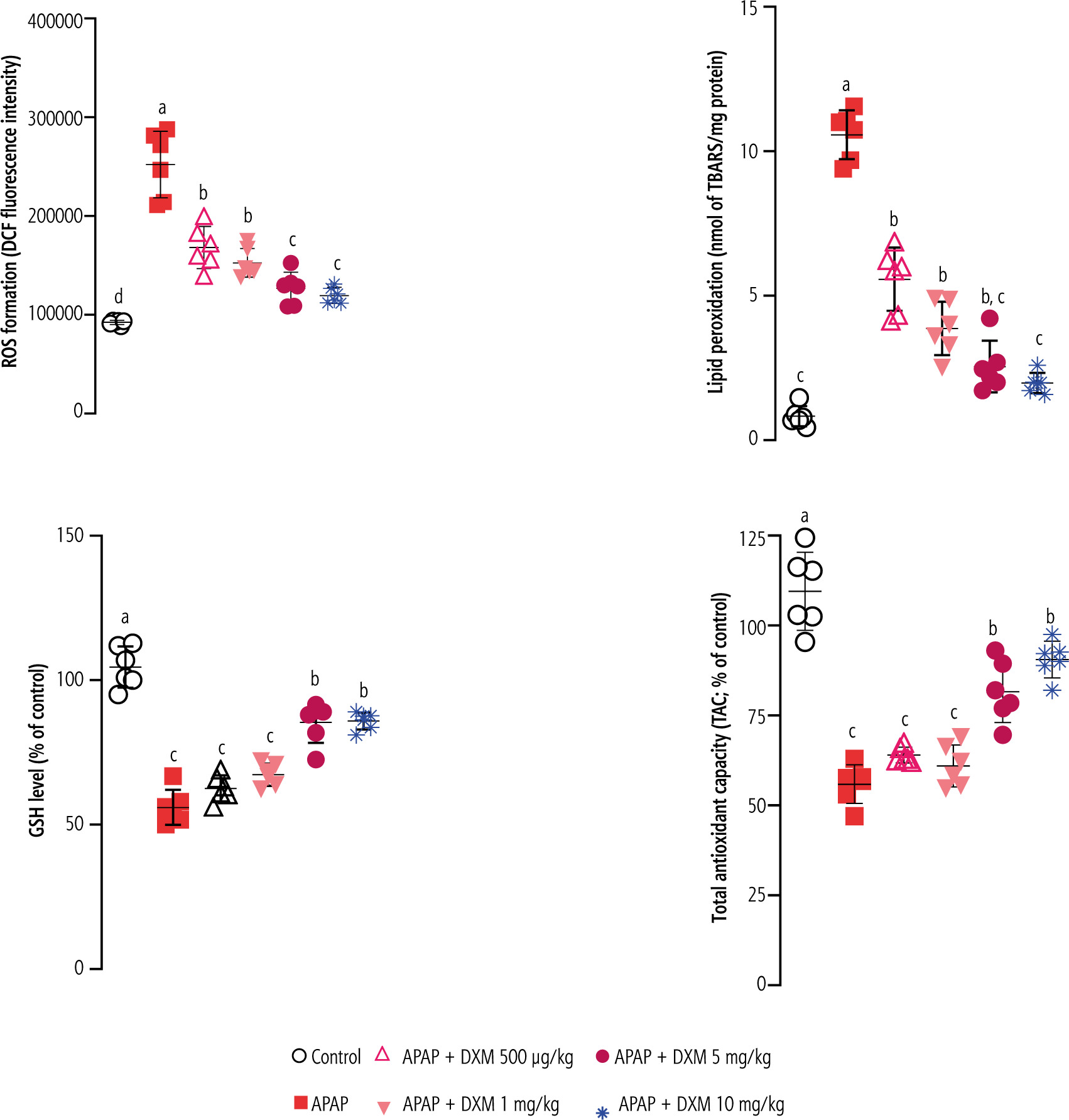
Significant ROS formation, lipid peroxidation, brain GSH depletion, and impaired antioxidant capacity were detected in APAP-treated mice. It was found that DXM significantly blunted biomarkers of oxidative stress in the brain of APAP-treated animals. The effect of DXM on markers of oxidative stress was more prominent at higher doses of this drug (5 and 10 mg/kg) (Fig. 3).
Fig. 3
Biomarkers of oxidative stress in the brain of acetaminophen (APAP)-treated animals. Dextromethorphan (DXM) significantly blunted oxidative stress and its associated complications in the current study. Data are expressed as mean ±SD (n = 6). Different alphabetical superscripts indicate that the mentioned groups are significantly different (p < 0.05)
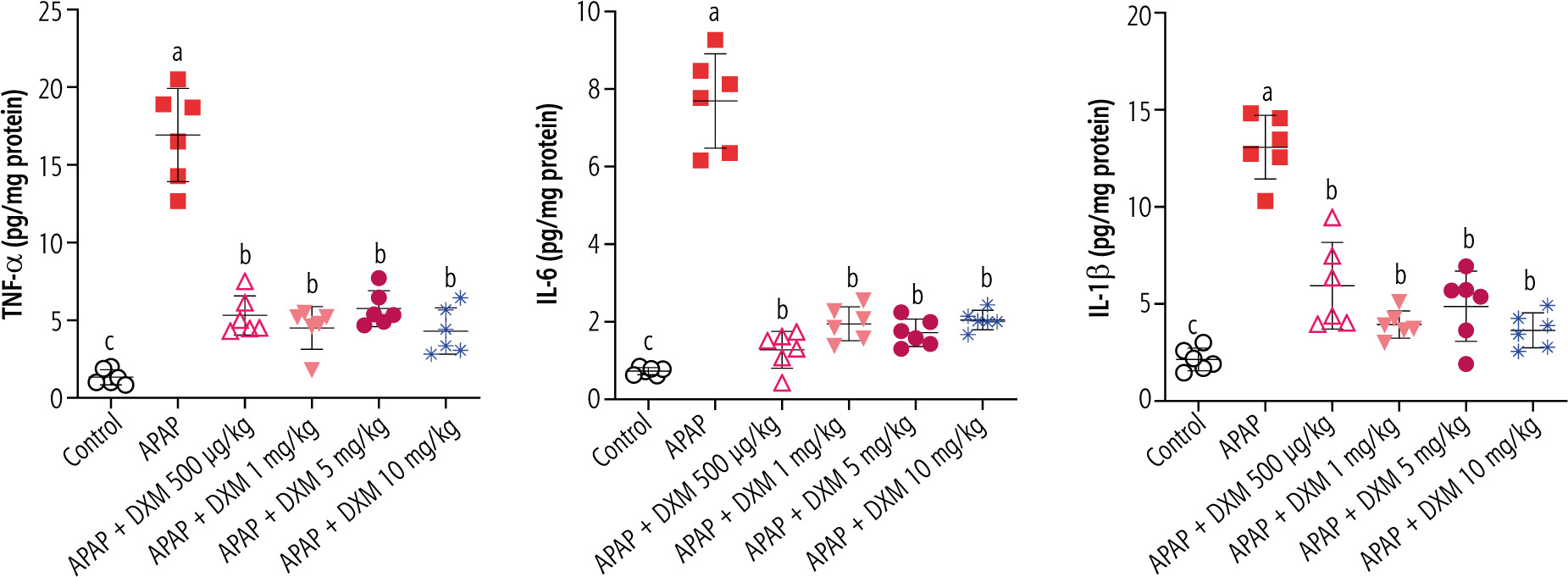
A significant increase in pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) was evident in APAP-treated mice compared to the control animals. It was found that DXM significantly decreased the level of pro-inflammatory cytokines in the brain tissue. No significant difference between the potency of DXM doses in decreasing brain cytokines was detected in the current study (Fig. 4).
Fig. 4
Effect of dextromethorphan on pro-inflammatory cytokine levels in the brain of hyperammonemic mice. APAP – acetaminophen. Data are expressed as mean ±SD (n = 6). Different alphabetical superscripts indicate that the mentioned groups are significantly different (p < 0.05)
No mortality was recorded in the current experimental model (24 h after APAP administration) in APAP and/or DXM-treated animals.
Discussion
Hepatic encephalopathy is a serious clinical complication that needs immediate and restricted therapeutic interventions. The ammonium ion (NH4+) is the major culprit responsible for HE-induced neurological disorders. Although there are several strategies for decreasing blood and brain NH4+ in HE, there is no specific pharmacological intervention to blunt HE-induced brain injury. The current study found that low-dose DXM significantly improved locomotor activity in an animal model of HE. Moreover, DXM significantly decreased oxidative stress biomarkers and proinflammatory cytokines in the brain of mice with HE. These data introduce DXM as a potential neuroprotective agent in the pharmacotherapy of HE-associated complications.
Increasing evidence indicates the pivotal role of oxidative stress and its associated complications in the pathogenesis of HE-induced CNS damage [3]. Therefore, agents which could stimulate cellular antioxidant mechanisms are potential neuroprotective agents in HE [3]. Interestingly, it has been found that DXM could significantly stimulate Nrf2 signaling in various experimental models [27]. Nrf2 signaling could induce the expression of a wide range of important cellular defense mechanisms such as superoxide dismutase, catalase, glutathione-s-transferase, and homoxygenase (HO-1) [67]. Hence, the activation of Nrf2 signaling by DXM could enhance cellular antioxidant capacity and decrease ROS levels. The current study found that DXM could significantly decrease ROS levels and enhance the antioxidant capacity of brain tissue in hyperammonemic animals (Fig. 3). Interestingly, it has been well documented that some enzymes expressed by Nrf2 signaling, such as HO-1, induce profound neuroprotective properties. HO1 induces the production of molecules such as biliverdin. It is well documented that biliverdin could act as a potential neuroprotective molecule against oxidative stress [68]. All these data indicate the important effect of DXM on oxidative stress biomarkers as a pivotal mechanism of action for the neuroprotective properties of this drug. Future studies on the effects of DXM on oxidative stress-related pathways (e.g., Nrf2) and the expression of related enzymes could enhance our understanding of its molecular mechanisms of action in conditions such as HE.
A plethora of studies have investigated the fundamental role of the inflammatory response and neuroinflammation in the pathogenesis of HE [16, 18, 19, 69, 70]. It has been repeatedly documented that the cerebral level of pro-inflammatory cytokines (e.g., TNF-α and IL-1β) are significantly increased in HE [71, 72]. The connection between increased brain pro-inflammatory cytokine level and oxidative stress has also been mentioned in previous investigations [4, 73, 74]. It has been found that neuroinflammation could deteriorate oxidative stress [18, 71, 75]. Astrocytes are major sources of inflammatory cytokines in ammonia neurotoxicity [76-78]. Meanwhile, inflammatory cytokines could trigger the accumulation of inflammatory cells such as neutrophils in the CNS [18, 71, 75]. Inflammatory cells such as neutrophils are primarily involved in ROS generation and oxidative stress [79]. All these data reflect the interplay between the inflammatory response and oxidative stress in ammonia neurotoxicity. On the other hand, the effect of DXM on the major cellular signaling mechanism involved in the inflammatory response is an interesting feature of this drug [80, 81]. It has been reported that DXM could significantly suppress the natural killer κB (NFκB) signaling pathway [80, 81]. NF-κB signaling is the primary path for the expression of genes involved in synthesizing inflammatory cytokines [80, 82]. In this context, several studies have shown that DXM significantly decreased cytokine release from inflammatory cells [25, 83]. In the current study, we found that DXM significantly decreased the brain level of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) in an animal model of HE (Fig. 4). These data indicate that the effects of DXM on major pathways involved in the inflammatory response (e.g., NF-κB) could play a major role in its neuroprotective properties in an HE model. Further studies on the expression of NF-κB components in the brain of DXM-treated animals could enhance our understanding of its precise neuroprotective mechanism in HE.
The antagonistic effect of DXM on N-methyl-D-aspartate (NMDA) receptors is an exciting feature of this drug [20, 28, 31, 84, 85]. Several studies have revealed that DXM could significantly inhibit NMDA receptors’ hyperactivation and its consequent events (e.g., severe oxidative stress) [28, 33, 86]. On the other hand, the pathogenic role of NMDA receptors’ hyper-activation in HE is widely investigated [3, 87]. NMDA hyperactivation and its consequent events in the CNS are known as excitotoxicity [3]. It is well known that excitotoxicity is associated with disturbed cytoplasmic Ca2+ ion levels, mitochondrial impairment, and severe oxidative stress in the brain during hyperammonemia and HE [3]. Although not investigated in the current study, the antagonistic effects of DXM on NMDA receptors could play a role in its neuroprotective properties in hyperammonemia.
The effects of DXM on the functional deficit and cognitive impairment have been repeatedly mentioned in various experimental models [88-90]. Our data are in line with evidence indicating the positive effects of DXM on locomotor and cognitive function observed in previous experimental models. In this context, it should be mentioned that previous investigations indicate that a higher dose of DXM (e.g., > 20 mg/kg in experimental models and > 100 mg/kg in human cases) caused significant motor disorders [88, 91]. Therefore, lower doses of DXM were applied in the current study. The results obtained from the current study indicate that lower doses of DXM are effective for the management of HE-induced CNS complications. This could provide clues for the application of DXM in clinical settings.
The role of DXM in blunting oxidative stress and its associated complications, as well as the effects of this drug on neuroinflammation, seems to play a pivotal role in its mechanism of neuroprotection. On the other hand, DXM has a great safety profile and could be administered in critically ill patients (e.g., HE). These data provide new insight into the neuroprotective properties of DXM during hyperammonemia. Therefore, repurposing DXM could provide a novel therapeutic option for HE management in clinical settings.





