
Introduction
MELAS syndrome (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes) is a genetically determined disease caused by mutations in mitochondrial DNA. The most common is A3243G point mutation of mtDNA encoding the leucine tRNA (MT-TL 1 gene) [1]. The first case suggesting the diagnosis of MELAS was described in 1975 [2] The syndrome mainly causes dysfunctions of the nervous system and muscles. It is also associated with several endocrine disorders, e.g. short stature, delayed puberty, or diabetes mellitus [1].
Case report
We present a patient of Department of Endocrinology and Diabetology, The Children’s Memorial Health Institute. The girl was born at 40 weeks’ gestation with birth weight of 3160 g (–1.0 SDS) and length 55 cm (2.5 SDS). The parents of the patient are Caucasian, unrelated, and not affected by any chronic or genetic disorders. Uncomplicated neonatal period and normal psychomotor development were observed. A 3-year history of headaches was described; however, neurological evaluation was performed, and the EEG showed no abnormalities. From the age of 9 years the girl has been under ophthalmological care due to retinitis pigmentosa. There was no family history of endocrine and mitochondrial diseases.
The patient was first admitted to the Department of Endocrinology and Diabetology, The Children’s Memorial Health Institute at the age of 11.5 years. Physical examination revealed short stature (height 127.7 cm; –2.95 SDS), asymmetry of the palpebral fissures (right < left), cranial nerve VI palsy, hypotonia, and symmetrical hyporeflexia. Due to an abnormal neurological examination, a brain MRI was performed. It demonstrated hyperintense, bilateral lesions in the corticospinal tract, thalamus, and cerebellar peduncles in T2-weighted imaging (Fig. 1). In laboratory tests, the blood and urine concentration of lactic acid and pyruvate acid were elevated (urine was tested by the method of gas chromatography–mass spectrometry – GC-MS) (Table I). Additionally, the patient was diagnosed with sensory-neural hearing loss. Suspecting mitochondrial disease, a muscle biopsy was carried out. Histologically, ragged red fibres (RRF) with a normal activity of cytochrome c oxidase (COX) were present, which strongly indicated MELAS syndrome. Nevertheless, a genetic evaluation did not detect the most common mutations in the MT-TL1 gene (point mutation 3243A>G) and MT-ND5 gene (12770A>G, 13084 A>T, 13094T>C, 13513G>A, 13514A>G, 13528A>G). Despite this, the treatment was initiated, which consisted of L-arginine (2 g b.i.d.), Coenzyme Q10 (100 b.i.d.), and L-carnitine (500 mg b.i.d.). During follow-up, the cranial nerve palsy and the asymmetry of the palpebral fissure resolved. In a recent neurological examination, ptosis and upper limb tremor was present.
Figure 1
Magnetic resonance imaging of the brain. T-2 – weighted imaging: hiperintensive changes in cerebellar peduncles
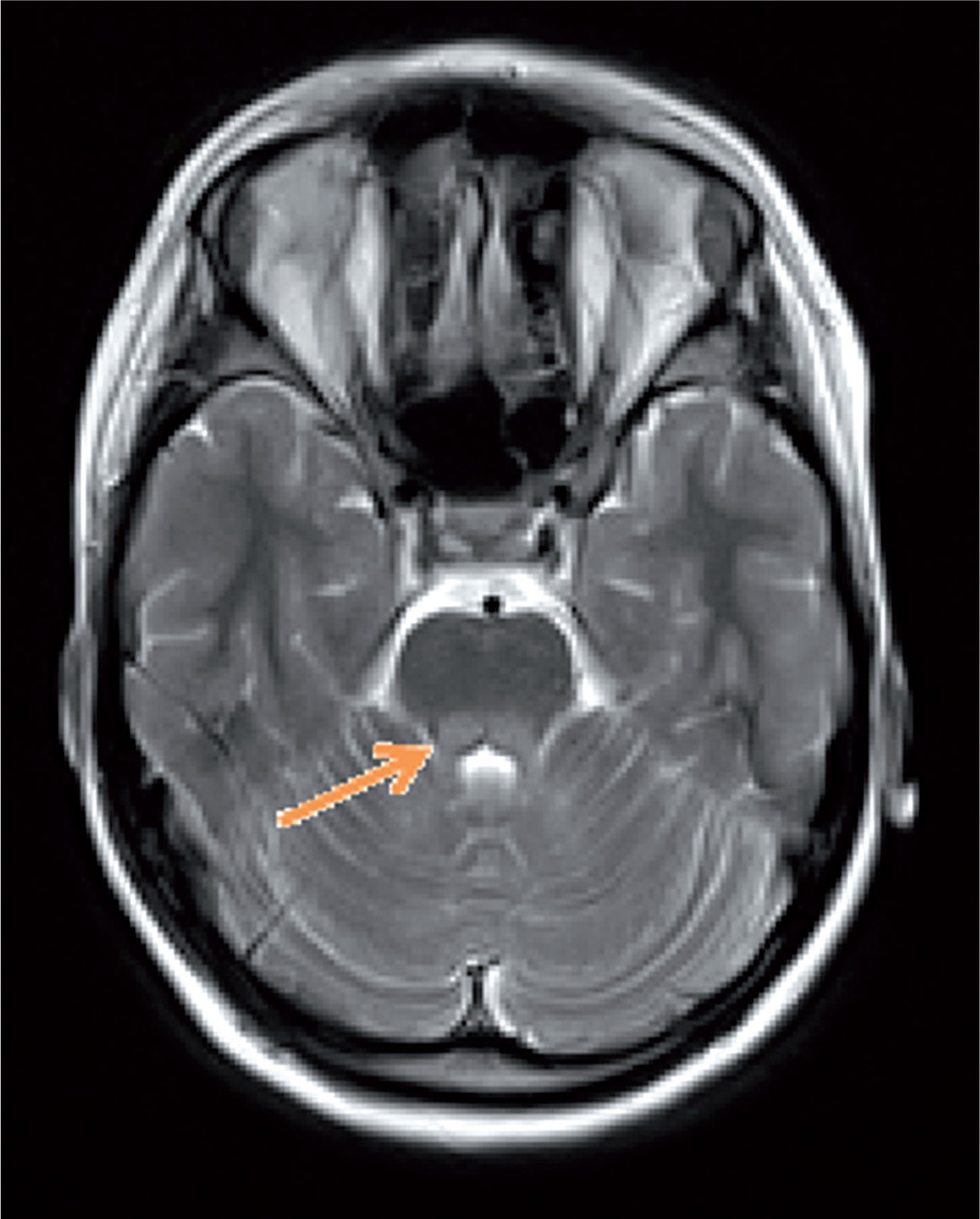
Table I
Laboratory tests results
[i] GC/MS – gas chromatography – mass spectrometry; IGF-1 – insulin-like growth factor 1; IGFBP3 – insulin-like growth factor-binding protein 3; TSH – thyroid-stimulating hormone; fT4 – free thyroxine; HbA1c – glycated hemoglobin A1c; anti-GAD – glutamic acid decarboxylase antibodies; anti-IA2 islet tyrosine phosphatase 2 antibodies; anti-ICA – islet cell antibodies
Concurrently, the patient underwent endocrinological diagnostics. Stunted growth has been observed from 8 years of age (Fig. 2). The peak level of growth hormone (GH) was 2.7 ng/ml, 2.8 ng/ml, and 2.8 ng/ml with a glucagon loading, with arginine and during sleep, respectively (Table II). Insulin-like growth factor-1 (IGF-1) value was also decreased, and bone age was delayed 2 years. Laboratory tests revealed no impairment of thyroid and adrenal gland function (Table I). GH deficiency was diagnosed, and recombinant human GH (rhGH) therapy was introduced at 0.59 U/kg/week, which resulted in growth acceleration from 4.2 to 6.5 cm/year. At the age of 14 years delay puberty was observed (Pub1, Th1, Ax1). The LH-RH test showed prepubertal values (Table II), bone age was estimated to be delayed by approximately 2.5 years. Due to the fact that first signs of puberty developed during surveillance, no hormonal therapy was introduced.
Figure 2
Percentil chart growth for children and adolescents in Poland 3-18 year of age. OLA and OLAF researches [3]
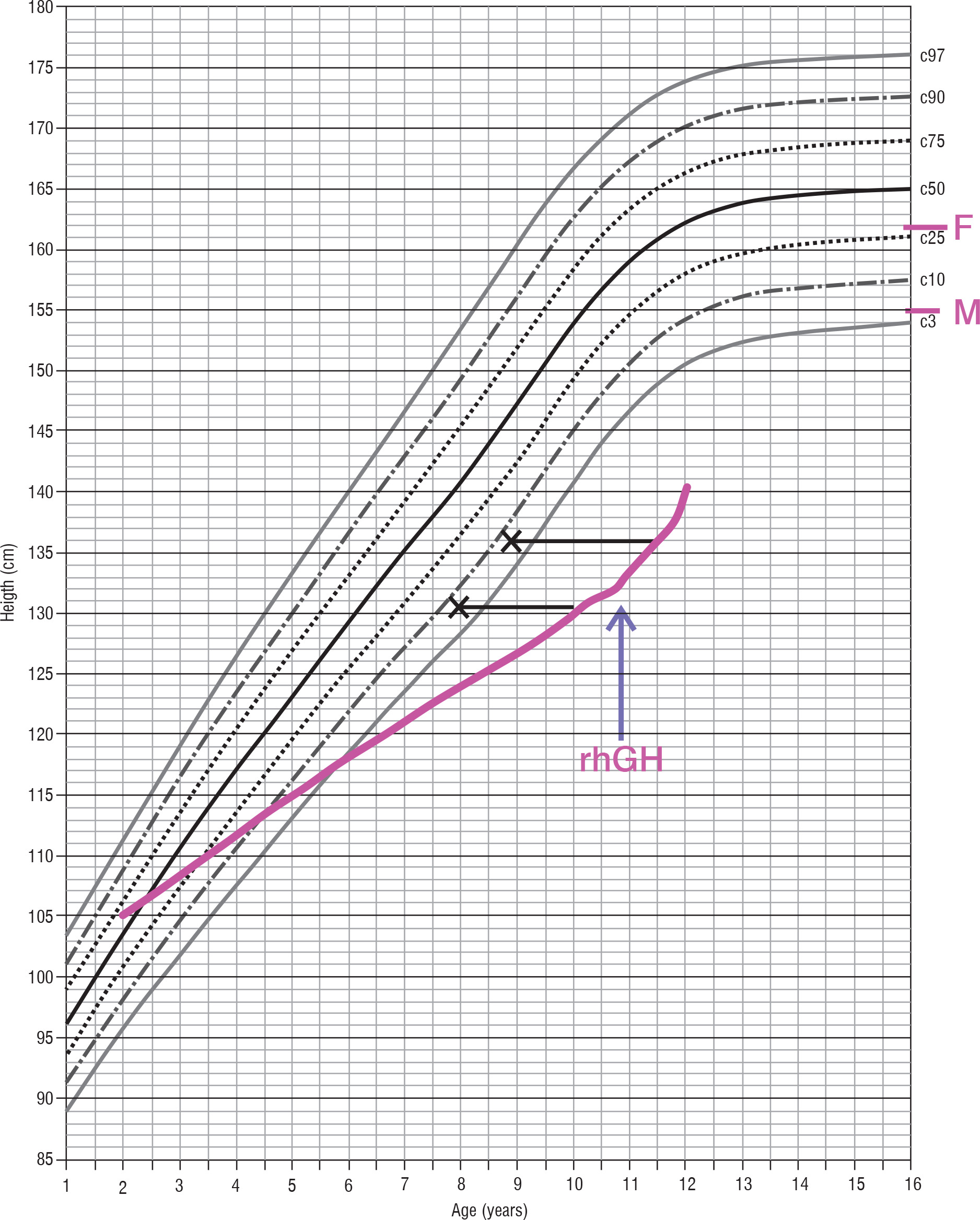
Table II
Tests results
After 1 year of the rhGH therapy a oral glucose tolerance test (OGTT) was performed which revealed diabetes. The increase of glucose level at the 120-minute time point and normal blood values of insulin were detected (Table II). Before the initiation of rhGH treatment, the OGTT was negative. Glycated haemoglobin A1c (HbA1c) before and during the rhGH therapy remained normal. Levels of glutamic acid decarboxylase antibodies (anti-GAD), islet tyrosine phosphatase 2 antibodies (anti-IA2), and islet cell antibodies (anti-ICA) were not elevated; therefore, diabetes type 1 was excluded (Table I). The patient started a carbohydrate exchange diet, achieving normoglycaemia. The treatment of rhGH was continued.
Discussion
MELAS syndrome is a genetically determined disease caused by mutations in mitochondrial DNA. The most common (about 80% of cases) is A3243G point mutation of mtDNA encoding the leucine tRNA (MT-TL 1 gene). In addition, other point mutations in the MT-TL 1 and MT-ND 5 gene are described. It is estimated that the mutations of mtDNA are not identified in up to 10% of patients with MELAS [1]. In our case we did not detect the most common mutations; nevertheless, this does not exclude the diagnosis of MELAS syndrome. Genetic analysis using a next-generation sequencing (NGS) method is awaited.
Clinical manifestation of MELAS is variable. The first symptoms may appear both in childhood and in young adults (only 5–8% of cases before 2 years old and 1–6% above 40 years old) [4]. According to several authors, the syndrome can be divided into juvenile and adult form depending on the onset of first symptoms [5].
Neurological symptoms, myopathy, and lactic acidosis are the 3 main manifestations of MELAS. Stroke-like episodes are the most common neurological disorders, characterized by the following: reversible aphasia, hemiplegia, cortical blindness, etc. Localized or generalized epilepsy can also occur, mostly as a result of the presence of ischaemic foci [5–7]. Typical brain imaging findings include stroke-like areas, bilateral abnormalities in the deep grey matter or brainstem basal ganglia, or cerebellar atrophy [8]. Similarly to our patient, the vast majority of patients suffer from recurrent headaches. Attention should be also paid to psychiatric symptoms such as depression, bipolar disorder, or even psychosis [5–7].
Another major symptom of MELAS is myopathy, which may present as a muscle weakness and exercise intolerance. Muscle biopsy is necessary, and it can reveal characteristic abnormalities, which are RRF. This pathology can be also seen in other mitochondrial cytopathies, but a large proportion of RRF with normal activity of COX may guide the way to MELAS diagnosis [1]. Lactic acidosis affects the majority of patients with MELAS; however, the normal lactic acid concentration does not exclude this diagnosis. Watchful monitoring of the metabolic status is very important in these patients, especially in a situation of active infection or during surgical procedures [9].
Patients with MELAS rarely suffer from cardiological problems (e.g. cardiomyopathy or Wolff-Parkinson-White syndrome). Other uncommon manifestations may include gastrological (cyclic vomiting, diarrhoea, constipation, recurrent pancreatitis), ophthalmic (retinitis pigmentosa), or audiological (sensorineural hearing loss) disorders [4–7].
Endocrine problems in patients with MELAS are another important issue. Short stature is observed in 33–82% of these patients [4–5]. Growth hormone deficiency has been described in multiple case reports [11–13].
It is presumed that in patients with mitochondrial diseases, the hypothalamic-pituitary-axis dysfunction is a potential mechanism of GH deficiency. Mutsuzaki et al. concluded that the dysfunction of the hypothalamic cells producing growth hormone-releasing hormone (GH-RH) is the initial impairment. They hypothesize that these cells are much more vulnerable to lactic acidosis than the anterior pituitary cells producing GH. Chronic ischaemia and energetic deficiency of hypothalamic cells is also postulated [13]. It also appears that the anatomical abnormality is not an issue, due to the fact that in most patients with MELAS, brain MRI does not show abnormalities of the pituitary gland and the hypothalamic area [11]. Notwithstanding, the small number of cases described and a variety of clinical features resulting from heteroplasmy of mitochondrial DNA does not allow a single hypothesis to be put forward. Despite the fact that most patients with confirmed GH deficiency receive rhGH treatment, not every individual will achieve an improvement in the rate of growth. Our patient reached an average growth acceleration. We did not notice any exacerbation of the course of mitochondrial disease during the observation period. In the literature, short-term follow-up does not reveal any significant adverse effects of the rhGH therapy in patients with MELAS; however, there are no data on the long-term safety of the aforementioned treatment. Therefore, a careful qualification and a close monitoring of rhGH therapy in the patients with mitochondrial disease is recommended [11, 13].
Approximately 1/3 of patients with MELAS are diagnosed with diabetes, with higher prevalence in adult form [5, 14, 15]. The average age of the onset of diabetes is 38 years [4, 16]. The mechanism of diabetes in MELAS syndrome remains unclear; nevertheless, several explanations are considered. A disturbance of glucose consumption in peripheral tissues can cause insulin resistance and insufficient insulin secretion (reduction of ATP concentration as a result of mitochondrial disease leads to a decrease in insulin release by ATP – sensitive potassium channels). In addition, the higher concentration of lactates found in MELAS causes an increase in gluconeo-genesis, for which lactic acid is a substrate [17]. In our patient the cause of diabetes remains unknown. It could be the effect of rhGH therapy or a component of MELAS, or both of them. Another complex issue is the treatment of diabetes related to mitochondrial diseases, because currently no specific guidelines are available. However, it is known that due to the risk of lactic acidosis, metformin is contraindicated. Thiazolidinedi-ones reduce hepatic gluconeogenesis and increase glucose utilization in muscles and adipose tissue, as well as preserving beta-cell mass. On the other hand, thiazolidinediones inhibit the mitochondrial purine carrier, which reduces the purine uptake in mitochondria and, in this way, they can increase the risk of lactic acidosis. Incretin drugs such as GLP-1 receptor agonists or DPP-4 inhibitors seem to be the best therapeutic choice. They increase the secretion of insulin from beta cells and also inhibit the glucagon secretion. They also have antioxidant and anti-inflammatory effects [17]. Currently, our patient is on a carbohydrate exchange diet, achieving normoglycaemia.
In MELAS syndrome, we can observe a hypogonadotropic hypogonadism with low level of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). In some patients a degree of pubertal development may occur, but oligomenorrhoea or secondary amenorrhoea can develop [12, 16].
There are also other endocrinological disorders in MELAS syndrome. We can mention secondary hypothyroidism (due to the hypothalamic dysfunction) or the case of a patient with primary adrenal insufficiency and MELAS syndrome [18].
Conclusions
MELAS is a mitochondrial multi-systemic disease, and therefore patients require multi-specialist care. Endocrine disorders may be one of the first manifestations of this syndrome. In differential diagnosis of short stature, less common causes such as mitochondrial diseases should be taken into consideration. Regular glycaemic control is recommended due to the risk of diabetes.









