
Introduction
Dyslipidemias can often present with cutaneous manifestations which are important pointers aiding early diagnosis, helping prevent later complications. Of these, xanthomas are important manifestations which may present in skin, tendons, ligaments, fascia and periosteum [1]. These cutaneous deposits of cholesteryl ester-enriched foamy macrophagesmay appear as single or multiple non-tender lesions which may be pea-sized or larger. Less commonly, dyslipidemias may present with cholesterol deposits in cornea, known as “arcus lipoides corneae” [2]. Herein, we present a child who presented with multiple xanthomas and arcus lipoides corneae and was subsequently diagnosed to have underlying familial hypercholesterolemia.
Case-report
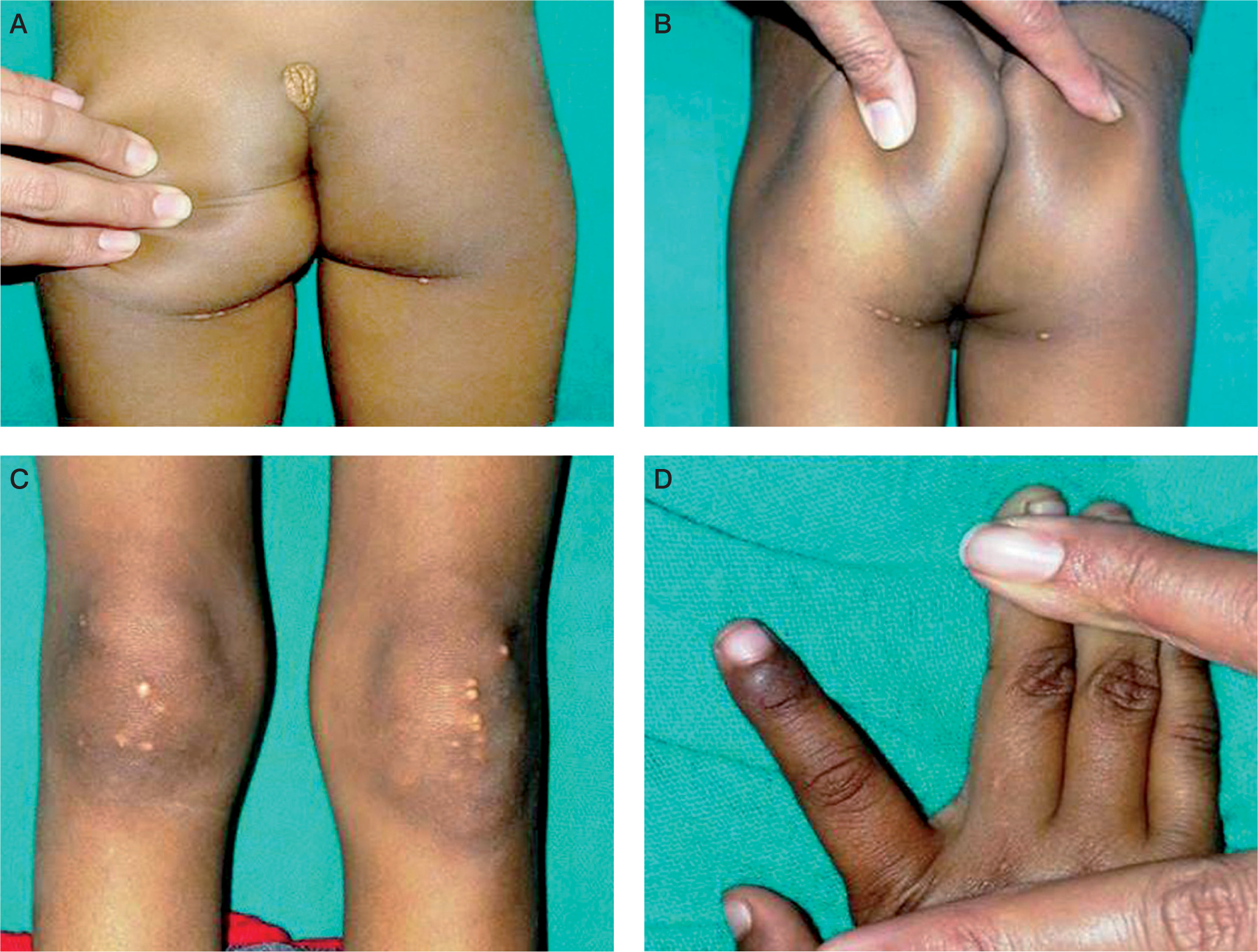
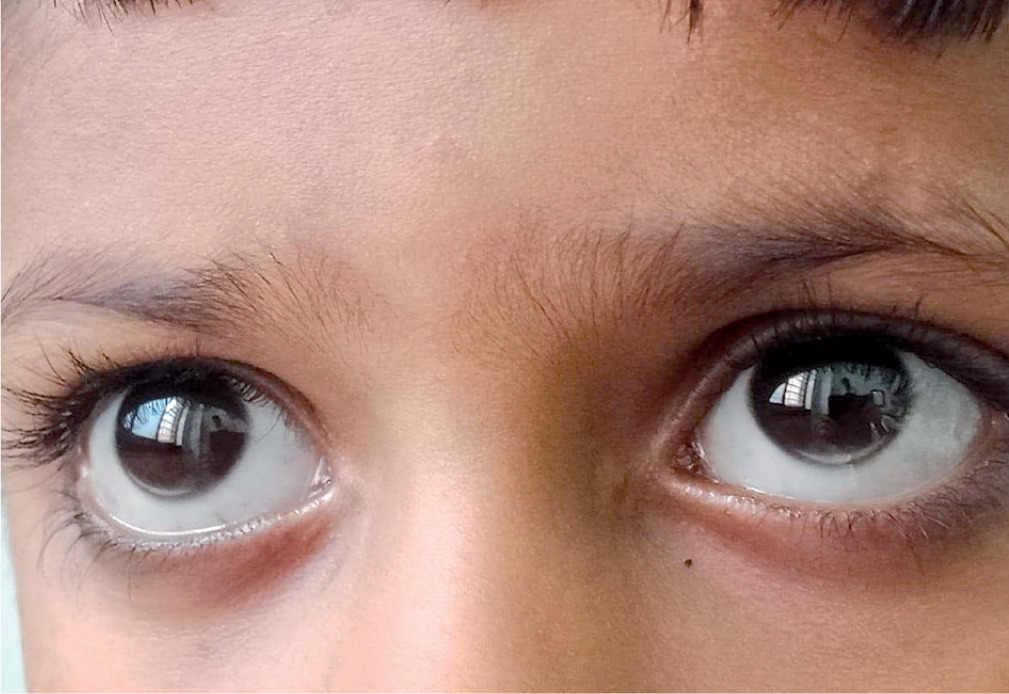
A 5-year-old boy presented with a tumorous lump over the lumbosacral region (Fig. 1A) and multiple yellowish white nodules over both buttocks (Fig. 1B) and knees (Fig. 1C). These had been increasing gradually in size and number over the last one year. General physical examination revealed multiple xanthomas over both gluteal regions, extensor aspects of both knees and in interdigital spaces (Fig. 1D). A whitish gray arc was also seen along the lower margin on the cornea (Arcus juvenilis) in both eyes (Fig. 2).
Figure 1
A) Tuberous xanthomas seen on lower back. B) Tuberous xanthomas seen on both buttocks. C) Tendinous xanthomas seen on extensor aspects of both knees. D) Interrdigital xanthomas
The child had a weight of 13 kg and height of 100 cm (weight for height Z score –1.79; Body mass index Z-score –1.72). Rest of the systemic examination and fundoscopy was unremarkable. A possible diagnosis of dyslipidemia was considered. Fasting lipid profile revealed hypercholesterolemia with normal serum triglyceride levels (Table I). Liver function test revealed serum glutamic pyruvic transaminase 65 IU/l, serum glutamic-oxaloacetic transaminase 71 IU/l, alkaline phosphatase 814 IU/l, bilirubin 0.4 mg/dl, total protein 7.3 g/dl, and serum albumin 3.7 g/dl. The thyroid function tests revealed serum thyroid stimulating hormone (TSH) 3.33 mIU/ml (reference: 0.44–3.77), free thyroxine (fT4) 0.91 ng/dl (reference 0.76–1.66), and free triidothyronine (fT3) 3.57 pg/ml (reference 2.15–4.12).The hemogram, kidney function test, blood sugar (fasting and post-prandial), urine analysis, electrocardiogram, carotid doppler and echocardiogram were unremarkable. Ultrasound of abdomen revealed mild hepatomegaly with fatty changes. The parents and one younger sibling aged one-year were also investigated for dyslipidemia. The mother was noticed to have multiple tendinous xanthomas with evidence of hypercholesterolemia; the father and younger sibling had a normal lipid profile. A diagnosis of familial hypercholesterolemia was made for the index case and his mother.
Table I
Serum lipid profile of the index case and his mother
The child and his mother were started on a low cholesterol diet and oral atorvastatin and were advised lifestyle modifications. After three months, the serum cholesterol levels for the child were 480 mg/dl and oral cholestyramine and nicotinic acid were added to his treatment.
Discussion
There are four general classes of pediatric dyslipidemias:
medication-related dyslipidemia,
dyslipidemia related to lifestyle factors,
genetic dyslipidemia, and
dyslipidemia secondary to medical disorders like diabetes and hypothyroidism [3].
Based on the concentrations of cholesterol and triglycerides in blood, three types of genetic dyslipidemia are recognized:
Familial hypercholesterolemia (FH): It is usually an autosomal dominant condition, characterized by high cholesterol concentrations and normal triglyceride concentrations due to decreased low-density lipoprotein cholesterol (LDL-C) clearance, as seen in our index case and his mother
Familial combined hyperlipidemia: It is characterized by a high LDL-C and triglycerides (TGs) which are a consequence of increased apolipoprotein B production. It also predisposes to risk for cardiovascular disease (CVD).
Familial severe hypertriglyceridemia (HTG): The most common genotype is homozygous autosomal recessive loss of function mutations in lipoprotein lipase or apolipoprotein C2 leading to significantly increased serum TG. Levels of TG > 1000 mg/dl are associated with life threatening pancreatitis.
Familial hypercholesterolemia is among the commonest dyslipidemias. Mutations in the genes related to the LDL receptor (LDLR) pathway including mutations in the apolipoprotein B (apoB) gene (APOB) or gain-of-function mutations in the proprotein convertase subtilisin-kexin type 9 [PCSK9] gene, result in FH which manifests with marked elevation of plasma LDL-C levels from birth. The patients with FH could be homozygotes (prevalence of 1 in one million) or heterozygotes (prevalence of 1 in 500). In the absence of treatment, total cholesterol and LDL-C levels in homozygous FH are usually 650 to 1000 mg/dl and > 500 mg/dl, respectively, while corresponding figures for heterozygotes with FH are 350–550 mg/dl and 220 mg/dl respectively [4].
Homozygotes frequently develop xanthalesma around the eyelids or cutaneous xanthomas on the extensor surfaces of joints and tendons, and rarely arcus lipoides corneae (arcus senilis/arcus juvenilis/arcus cornealis) in the eyes. Xanthomas are cutaneous deposits of foamy macrophages laden with cholesterol, while the arcus lipoides corneae is believed to be a consequence of increased endothelial permeability of the limbal vasculature to lipids leading to increased deposition of cholestrol in the cornea. It is referred to as “arcus juvenilis” in children and young adults or as “arcus senilis” in older adults [2]. While xanthomas are more common in FH, arcus lipoides corneae is an uncommon finding in children with FH [5, 6] and is seen more commonly in older patients [7]. Children and adolescents with homozygous FH are particularly predisposed to myocardial infarction and ischemic cardiomyopathy, often in the first 2 decades of life. The heterozygotes also have a predilection for early atherosclerosis.
Diagnosis of FH can be established using any of the three criteria exist, viz, the Dutch Lipid Clinic Network [7], the US MedPed (Make Early Diagnosis To Prevent Early Death) program [8], and the Simon Broome Register Group in the UK [9], which rely on a combination of parameters based on serum cholesterol levels, family history of dyslipidemia, clinical examination findings like the presence of xanthomas or arcus juvenilis, and genetic testing. Our child was diagnosed as FH based on the criteria laid by the Dutch Lipid Clinic Network [7] as he scored a total of 20 points on this scale (total score >8 is diagnostic of FH) as follows:
score based on family history (First degree relative with tendon xanthomata): 2,
score based on physical examination (Tendon xanthomata: 6, arcus cornealis at age <45y: 4): 10,
score based on laboratory assessment (LDL-C>330 mg/dl with normal HDL-C and triglyceride levels): 8.
He also met the diagnostic criteria for FH as per the US MedPed (Make Early Diagnosis to Prevent Early Death) program [8]. Genetic testing for FH is not always warranted and absence of genetic markers does not exclude FH. DNA analysis for functional mutations in the LDLR, APOB, and PCSK9 can help establish the diagnosis of FH with certainty but are often costly and may not be readily available. Timely detection of dyslipidemias is particularly important as children can be advised for dietary and lifestyle modification in addition to pharmacotherapy to possibly decrease the risk for CVD. However, the treatment of FH in young children can be particularly challenging as the safety and efficacy of statins in children below 10 years is not clearly established and treatment modalities like weekly plasmapharesis may not be feasible [10]. Bile acid sequestrants and anti-oxidant supplements have been offered additionally. In the absence of adequate therapeutic modalities, key to management of FH is early diagnosis and lifestyle interventions and screening for CVD.









