
Introduction
Renal cell carcinoma (RCC) develops from the epithelial cells of renal tubules, accounting for more than 90% of kidney malignancies and approximately 4.4% of all malignant tumors. It ranks 14th in global incidence [1, 2]. Recently, RCC cancers of unknown primary (CUP-RCC) have emerged as a distinct subset of CUP. Sharing morphological and molecular features with RCC, CUP-RCC is treated with RCC-specific therapies, contributing to the observed increase in RCC incidence due to advances in diagnostics [3]. Mortality from RCC constitutes 1.6% of all cancer-related deaths [1, 2]. It is estimated that, at the time of diagnosis, metastatic RCC is present in approximately 17% of patients [4]. However, in recent years, continuous advancements in medical diagnostic technology have contributed to heightened rates of early RCC detection and favorable clinical prognoses. Exosomes have emerged as promising non-invasive tumor biomarkers. Their bilayer membrane protects enclosed mRNAs, miRNAs, and proteins, making them sensitive markers for diagnosis. Tumor-derived exosomal miRNAs in serum and urine show potential for early detection and monitoring in RCC [5] An increase in incidence is observed among individuals over 60 years of age and in males [2, 4].
In 2022, the World Health Organization (WHO) published the 5th edition of the Classification of Urinary and Male Genital Tumors, which is based on current knowledge regarding the morphological, immunohistochemical, molecular, epidemiological, and clinical characteristics of individual kidney tumors [6].
Surgical treatment can provide a cure for RCC if performed on individuals with localized disease; however, recurrence can occur in a subset of patients [7]. Adjuvant treatment should be considered for patients with clear cell RCC who are at high risk of recurrence following radical surgical treatment [8, 9]. Renal cell carcinoma is characterized by chemo- and radioresistance, which has unfavorably impacted the chances of a cure, particularly in advanced disease. A breakthrough in therapy occurred with the introduction of molecularly targeted treatments aimed at specific intracellular signaling pathways. These include tyrosine kinase inhibitors (TKIs) and inhibitors of the mammalian target of rapamycin (mTOR) pathway [7]. Recent studies have shown that downregulation of lactotransferrin (LTF), a key protein in the innate immune system, promotes metastasis in clear cell RCC (ccRCC). Interestingly, this downregulation also increases the responsiveness of ccRCC tumor cells to mTOR inhibitors, suggesting that LTF levels could serve as a predictive biomarker for the effectiveness of mTOR-targeted therapies [10]. Another treatment option for RCC is immune checkpoint inhibitor therapy (i.e., immunotherapy), which involves the use of monoclonal antibodies targeting programmed cell death protein 1 and cytotoxic T-lymphocyte antigen 4 [11]. Currently, the aforementioned drugs are used in monotherapy or combination therapy [12].
The aim of this study was to review and analyze the available literature on the interactions between TKIs used to treat advanced RCC and commonly consumed food products and dietary supplements.
Kinase inhibitors used in renal cancer
One of the main treatment strategies for advanced RCC is the use of TKIs. Tyrosine kinases (TK) belong to a group of enzyme proteins (protein kinases) that transfer a phosphate group from a phosphate donor to a substrate – an amino acid acceptor [13]. There are 4 groups of protein kinases: serine/threonine, tyrosine, histidine, and asparagine/glutamine kinases [14]. There are also dual-specificity kinases that phosphorylate serine, threonine, and tyrosine residues. Protein kinases play crucial roles in regulating essential cellular processes such as cell division, differentiation, and apoptosis [15]. Stimulation of cell division occurs primarily through interactions between growth factors and receptors located on the cell membrane. Most of these receptors have a TK activity domain, which is located in the cytoplasm. Examples of ligands for these receptors include growth factors such as vascular endothelial growth factor (VEGF), platelet-derived growth factor, and insulin-like growth factor. The loss of functional von Hippel-Lindau protein drives ccRCC via hypoxia-inducible factor 2-α (HIF-2α) accumulation and constitutive HIF activity. Belzutifan, a HIF-2α inhibitor, blocks its dimerization with HIF-1β, showing efficacy in ccRCC. A phase III trial reported belzutifan’s superiority over everolimus in progression-free survival (PFS) and objective response rates in advanced ccRCC [16]. The action of growth factors on receptors activates receptor kinase activity, triggering signaling pathways that result in changes in gene expression patterns, leading to mitosis. The deregulation of these pathways contributes to the formation of cancer cells through the overexpression of growth factors and receptors. Mutations in the cytoplasmic domain of TK also have an impact on this [17]. Abnormal protein kinase activity is the main mechanism by which cancer cells evade the physiological mechanisms that limit growth and survival [18]. Figure 1 illustrates the mechanism of action of TKIs.
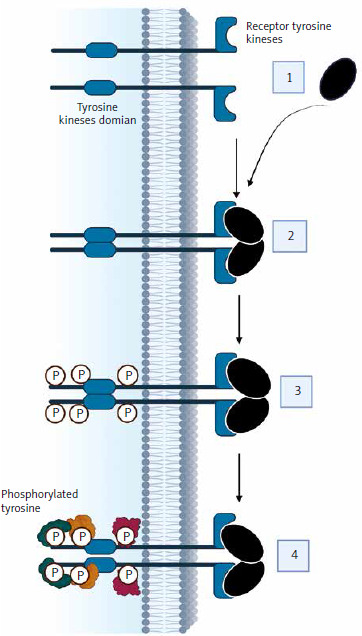
In the various stages of advanced RCC therapy, the following TKIs are utilized: sunitinib, pazopanib, cabozantinib, lenvatinib, axitinib, sorafenib, and tivozanib [8]. TKIs such as sunitinib, pazopanib, cabozantinib, lenvatinib, axitinib, sorafenib, and tivozanib are integral to advanced RCC treatment. For patients ineligible for immunotherapy, TKI monotherapy remains a suitable first-line option. The STAR trial specifically evaluated the efficacy of TKI monotherapy in this cohort. Additionally, some patients may receive TKIs as second-line monotherapy following other treatments. While treatment breaks can be considered to mitigate toxicity, caution is advised, as these patients often experience shorter PFS compared to those on continuous first-line TKI therapy [19]. Table 1 presents the characteristics of these drugs.
Table 1
Characteristics of tyrosine kinase inhibitors used to treat advanced renal cell carcinoma, approved by the European Medicine Agency and the Food and Drug Administration (based on product characteristics, 15.10.2024)
| Inhibitor | Mechanism of action | References |
|---|---|---|
| Axitinib | Inhibits RTKs: | [8, 9] [20, 21] |
| Cabozantinib | Inhibits tyrosine activity of: | [8, 9] [20, 22] |
| Lenvatinib | Inhibits RTKs: | [8, 9] [20, 23] |
| Pazopanib | Inhibits RTKs: | [8, 9] [20, 24] |
| Sorafenib | Multikinase inhibitor: | [8, 9] [20, 25] |
| Sunitinib | Inhibits RTKs: | [8, 9] [20, 26] |
| Tivozanib | Inhibits RTKs: | [8, 9] [20, 27] |
[i] c-Fms – transmembrane glycoprotein receptor tyrosine inhibitor, CSF-1R – colony stimulating factor receptor, FGFR – fibroblast growth factor receptor, FLT3 – Fms-like tyrosine kinase-3, GAS6/AXL – growth arrest-specific protein 6/receptor – member of the tumor-associated macrophage family tyrosine kinase receptors, Itk – interleukin-2 receptor inducible T-cell kinase, KIT – stem cell growth factor receptor, Lck – leukocyte-specific protein tyrosine kinase, MET – mesenchymal epithelial transition receptor, MER – receptor member of the tumor-associated macrophages family tyrosine kinase receptors, PDGFR – plated-derived growth factor receptor, RAF – V-raf murine sarcoma viral oncogene, RET – rearranged during transfection receptor, ROS1 – proto-oncogene 1 receptor, RTKs – receptor tyrosine kinases, TIE-2 – angiopoietin-1 receptor, TRKB – tropomyosin receptor kinase B, TYRO-3 – member of the tumor-associated macrophage family tyrosine kinase receptors, VEGFR – vascular endothelial growth factor receptor
TKIs used to treat advanced RCC are available orally. This formulation enhances the comfort of patient treatment, among other things, by allowing flexibility in the location and timing of medication intake. The treatment outcome is influenced not only by genetic factors but also by external variables such as dietary habits (e.g., timing and composition of meals), adherence to prescribed medication schedules, and the concomitant use of other substances, including food or drugs [28]. While TKIs offer flexibility in administration, they can cause treatment-mediated toxicities due to their inhibition of multiple targets. The extended presence of troublesome side effects could result in stopping the treatment [29]. Notably, hematologic and hepatic toxicities are significant and require careful monitoring. Although a definitive link between adverse event severity and treatment efficacy is lacking, TKIs’ blockade of VEGF receptors is associated with hypertension and other cardiovascular complications. Specifically, pazopanib may induce side effects such as hair depigmentation, diarrhea, nausea, anorexia, vomiting, and, in rare instances, posterior reversible encephalopathy syndrome [30].
Drug-food interactions
A drug is a chemical substance intended to induce a specific pharmacological effect once delivered to the body [31]. After administration, it undergoes a series of transformations, categorized into three types: pharmaceutical, pharmacodynamic, and pharmacokinetic. The pharmacokinetic phase is further divided into stages: release, absorption, distribution, metabolism, and excretion [32]. Similarly, the food we consume undergoes transformations and consists of a mixture of various chemical compounds. For this reason, there are significant opportunities for interactions between food components and drugs [33].
An interaction refers to the physical, chemical, or physiological influence between at least two substances, resulting in changes to their individual properties or in a combined effect [34]. Drug-food interaction specifically refers to changes in the strength, timing, mechanism of action, or toxicity of a medication caused by a food component [33]. Such interactions can lead to treatment failure, increased therapy toxicity, or worsening of nutritional status [35]. Knowledge about possible drug-drug interactions is fairly widespread, with numerous publications on the topic. However, drug-food interactions represent a relatively new area of medical research, and assessing the impact of diet and nutritional status on drug efficacy remains challenging. This challenge is due to the emergence of new therapeutic substances and the continuous diversity and evolution of dietary patterns [36].
Since 2017, within the framework of the European Cooperation in Science and Technology, an international research group called the European Network on Understanding Gastrointestinal Absorption-related Processes has been active, bringing together scientists from various fields. Their task is to analyze drug-food interactions and prepare appropriate recommendations [37]. Interactions between drugs and food can occur at any phase and stage of drug metabolism [38].
Pharmaceutical phase
The pharmaceutical phase pertains to the stage of preparing a drug for administration to the patient [39]. This phase takes place outside the body, so interactions between the drug and food are rare at this stage. An exception occurs when the drug and food/drink are combined before being administered to the patient, for example, through a gastric tube [37]. This interaction most commonly involves drug inactivation due to processes such as hydrolysis, oxidation, or neutralization [40].
In the case of TKIs used to treat advanced RCC, the tablet should not be crushed before administration and should be taken with chemically neutral water [21–27, 40].
Pharmacodynamic phase
The changes that occur in the body under the influence of drugs and the mechanisms of these changes are determined by the pharmacodynamic phase [41]. Pharmacodynamic drug-food interactions occur when the effect of a drug is altered by food. A positive action is called synergy, whereas an opposing action is called antagonism. Synergy involves a mutual increase in the effect of at least two substances taken at the same time or in close succession. On the other hand, antagonism is a reaction contrary to synergy [42, 43]. The pharmacodynamic phase determines clinical effects such as increased or decreased bioavailability, which can affect drug toxicity [35]. In the case of TKIs used in advanced RCC treatment, drug-food interactions occurring in the pharmacodynamic phase are poorly known and documented. This may be due to a lack of clear methods for evaluating interaction events in this phase [44]. Moreover, due to the increasing number of food ingredients, determining pharmacodynamic interactions is challenging [45].
Pharmacokinetic phase
The pharmacokinetic phase describes the fate of a drug in the body [42]. First, ingested food alters the physiological conditions in the gastrointestinal tract, impacting various stages of the pharmacokinetic processes of orally administered drugs [45]. Food has the greatest effect on pharmacological substances during the absorption stage (approximately 60%), followed by metabolism (27.14%) and elimination (12.86%) [46].
The release stage involves the drug transitioning into solutions within body fluids [39]. It consists of three parts: breakdown of the drug formulation, dissolution of the active substance, and transfer of the drug to the absorption site [42]. At this stage, drug-food interaction does not occur [40]. A critical condition for drug release and dissolution is an adequate volume of fluid in the gastrointestinal tract [45].
Although drug-food interaction typically does not occur at this stage [40], other factors, such as the use of laxatives or changes in gastrointestinal motility and fluid volume, may influence the release and dissolution of the drug. An adequate volume of fluid in the gastrointestinal tract remains a critical condition for drug release and dissolution [45, 47–50].
Absorption
Absorption is the transition of a medicinal substance into the systemic circulation. This stage is associated with the concept of bioavailability, which determines the amount of drug that reaches the bloodstream. For drugs administered intravenously or intra-arterially, the bioavailability is 100%. However, for orally administered drugs, this percentage is less than 100% and depends on various factors including the size of the drug molecule, its solubility in fats, the degree of drug ionization, and blood flow at the absorption site [51]. The bioavailability of drugs is evaluated on the basis of the biopharmaceutics drug disposition classification system, introduced in 2005. Drugs are categorized into 4 classes on the basis of their solubility and ability to pass through the gastrointestinal mucosa [51, 52]. Class 1 drugs are characterized by high solubility and high permeability. Class 4 drugs have low solubility and low permeability. The classification of a drug within a particular group allows the prediction of the range of interactions between the drug and another substance [53–55]. These groups are significant in the case of consuming a high-fat meal before taking the medication or in combination with it. Such a meal increases intestinal permeability, leading to higher drug concentrations in the plasma, which may increase drug toxicity [28].
Pazopanib, cabozantinib, axitinib, and sorafenib belong to group 2, whereas sunitinib belongs to group 4. However, there are no clear data on whether tivozanib and lenvatinib belong to group 2 or group 4 [28].
The absorption of orally administered medication usually begins in the oral cavity. This process then continues in the stomach and the small intestine, where it proceeds most rapidly due to the rich blood supply of the mucosa in this area of the gastrointestinal tract [41]. Food can affect bioavailability by accelerating, delaying, or increasing the absorption of the drug. It may also have no effect at all [43].
In the case of possible drug-food interactions, the type of meal/liquid consumed prior to taking the medication is significant at this stage. The type of food affects the motility of the gastrointestinal tract, which varies in different parts of the digestive system, thereby influencing the degree of dissolution and absorption of the drug. Since 2002, the US Food and Drug Administration has required that any application for the registration of a new substance assess the drug’s bioavailability on the basis of standardized guidelines, depending on the type of meal taken with it, and that this information be included in the Summary of Product Characteristics and Package Leaflet. The volume of fluid in the gastrointestinal tract also impacts drug absorption and bioavailability. It is advisable to wash down the medication with 150–220 ml of water [45], which dilutes digestive juices and thus reduces their negative impact on the drug [41].
In certain situations, when TKIs are taken, it is permissible to swallow the medication and wash it down with a small amount of nonfat yogurt – this possibility has been approved for nilotinib [56]. These data may be extrapolated to other small-molecule TKIs [28]. However, there is a lack of evidence that fluids other than water do not affect the reduction in bioavailability.
The type of meal or the timing of its consumption does not affect the absorption of axitinib [21], lenvatinib [23], sunitinib [26] or tivozanib [27]. Just before pazopanib [24] and sorafenib [25] are taken, high-fat meals are recommended. In the case of sorafenib, a high-fat meal decreases its bioavailability, which in turn reduces its effectiveness [25]. However, pazopanib accelerates its absorption, thereby increasing the risk of toxicity [24].
Accelerated absorption of cabozantinib has been demonstrated after a high-fat meal; however, no definitive information has been obtained regarding an increase in toxicity. Therefore, cabozantinib should not be taken with food [22]. In Table 2, the guidelines for the administration of TKIs in RCC are presented.
Table 2
Guidelines for administration of tyrosine kinase inhibitors in renal cell carcinoma
| Name of active substance | Application | Biopharmaceutical group number | References |
|---|---|---|---|
| Axitinib | With meal or without meal | 2 | [21, 28, 57] |
| Cabozantinib | The medication should be taken at least 1 hour before a meal or 2 hours after a meal. | 2 | [22, 28, 57] |
| Lenvatinib | With meal or without meal | 2 or 4 | [23, 28, 57] |
| Pazopanib | The medication should be taken at least 1 hour before a meal or 2 hours after a meal.* | 2 | [24, 28, 57] |
| Sorafenib | The medication should be taken at least 1 hour before a meal or 2 hours after a meal.* | 2 | [25, 28, 57] |
| Sunitinib | With meal or without meal | 4 | [26, 28, 57] |
| Tivozanib | With meal or without meal | 2 or 4 | [27, 28, 57] |
For absorption, the pH prevailing in different parts of the digestive tract is also important. Changes in pH in the digestive tract depend on the type and volume of food consumed, as well as on the amount and type of enzymes secreted into the digestive tract lumen for this purpose. Drugs undergo ionization above or below a certain pH value. Changes in pH affect the solubility of a drug as well as its absorption and bioavailability. TKIs belong to the group of weak bases [28].
Drugs in the bloodstream are transported passively – on the basis of concentration gradients – or actively through transporters [33, 42]. Transporters are proteins located primarily in the cell membrane of enterocytes and liver cells. They regulate the absorption and bioavailability of drugs [35]. The group of transporters includes organic anion-transporting polypeptides (OATPs), P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), oligopeptide transporter and multidrug resistance-associated proteins (MRPs). Drug molecules and nutrients often compete for the same transport pathways, leading to interactions between them and with transporters [45]. Both drugs and food components can act as modulators or substrates for specific transport proteins, thereby affecting the absorption and distribution processes of other substances [28].
TKIs used in RCC can be inhibitors, inducers, or substrates for specific transport proteins. Inhibitors decrease the concentration of a given substance in the blood, inducers increase the concentration, and substrates block the action of the transporter. These interactions most commonly involve P-gp and BCRP [58]. Cabozantinib and sorafenib are P-gp inhibitors [16, 17, 20]. Pazopanib is an OATP inhibitor [19] and tivozanib a BCRP inhibitor [21]. However, MRP inhibitors increase plasma concentrations of cabozantinib [22], whereas inhibitors of P-gp and BCRP increase plasma concentrations of pazopanib [24]. Inducers of P-gp and BCRP reduce the plasma concentration of pazopanib [24]. There is no relationship between sunitinib and endothelial transporters [26].
The consumption of grapefruit juice, orange juice, apple juice, and green tea inhibits the action of OATP [45]. Furanocoumarins, found in various vegetables, including those from the legume and celery families [59], as well as flavonoids [60], constitute a significant group of P-gp inhibitors. The most well-known inducer of P-gp is St. John’s wort (Hypericum perforatum) [45]. Additionally, flavonoids are also inhibitors of BCRP and MRPs [45].
Distribution
Distribution is the process of drug movement between compartments [61]. To exert its effects, a drug must reach its site of action at the appropriate concentration. Factors influencing distribution include the rate of diffusion, the drug’s affinity for tissues, blood flow in the area, and the binding of the drug to plasma proteins [51]. After entering the bloodstream or lymphatic system, drugs reversibly bind to circulating proteins, such as albumin, α-1 acid glycoprotein (AAG), and lipoproteins [61]. Albumin plays a major role in this process [45]. A drug that is bound to a protein is inactive and does not interact with cells. The binding of the drug to plasma proteins is reversible [51]. If the unbound drug is eliminated, some of the bound drug will detach from the plasma protein and be released [45].
A food ingredient can compete for the same binding site on plasma proteins as a drug. This may lead to changes in the distribution of the drug and its bioavailability [61]. The tyrosine kinase inhibitors used in RCC bind to plasma proteins, mainly albumin, in more than 95% of cases [21–27].
In the literature, data indicating that the interaction between drugs and nutrition is significant at this phase of pharmacokinetics are lacking. However, the nutritional status of the patient is important. Malnutrition can decrease the levels of albumin and AAG, which may lead to increased drug toxicity, whereas a high-protein diet can increase the concentration of proteins in plasma [45].
Metabolism
Metabolism is a process in which substances introduced into the body undergo chemical changes [62]. This process is divided into two phases: in phase 1, a group of enzymes modifies the chemical structure of the drug, and in phase 2, another group of enzymes converts these compounds into polar forms to facilitate their excretion. Drug metabolism primarily occurs in the liver but also occurs in the intestines, kidneys, and lungs [51]. Aging brings about unavoidable changes, including alterations in drug metabolism in the liver [63].
The enzyme groups responsible for drug metabolism are as follows: cytochrome P450 (CYP) – 73%, UDP-glucuronosyltransferases (UGTs) – 15%, esterases – 9%, flavin-containing monooxygenase (FMO), N-acetyltransferase (NAT) – 1%, and monoamine oxidase (MAO) – 1% [61]. Enzymes from the CYP450 and UGT [21–27] groups are involved in the metabolism of TKIs. Table 3 presents the enzymes involved in the metabolism of TKIs used in advanced kidney cancer.
Table 3
Enzymes involved in metabolism of tyrosine kinase inhibitors used in renal cell carcinoma
| Name of active substance | Metabolizing enzymes | References |
|---|---|---|
| Axitinib | Primarily metabolized by CYP3A4/5, minor contributions from CYP1A2 and CYP2C19. Also undergoes glucuronidation by UGT1A1. | [21, 57, 64, 65] |
| Cabozantinib | Metabolized mainly by CYP3A4, oxidation enhanced by cytochrome b5. Additionally, it is a substrate of MRP2. | [22, 57, 65] |
| Lenvatinib | Metabolized primarily by CYP3A4 and by AO. Non-enzymatic processes also contribute to its metabolism. | [23, 57, 65] |
| Pazopanib | Metabolized predominantly by CYP3A4, minor contributions from CYP1A2 and CYP2C8. Pazopanib inhibits UGT1A1-mediated glucuronidation. | [24, 57, 65] |
| Sorafenib | Metabolized primarily by CYP3A4 and undergoes glucuronidation by UGT1A9 | [25, 57, 65] |
| Sunitinib | Metabolized mainly by CYP3A4 | [26, 57, 65] |
| Tivozanib | Metabolized by CYP3A4 and CYP1A1. Also undergoes glucuronidation by UGT1A1, UGT1A3, UGT1A7, UGT1A8, UGT1A9, and UGT1A10. | [27, 57] |
[i] AO – aldehyde oxidase, CYP3A4 – cytochrome P450 3A4, CYP3A4/5 – cytochrome P450 3A4/5, CYP1A2 – cytochrome P450 1A2, CYP2C19 – cytochrome P450 2C19, CYP2C8 – cytochrome P450 2C8, CYP1A1 – cytochrome P450 1A1, MRP2 – multidrug, resistance-associated protein 2, UGT1A1 – uridine diphosphate glucuronosyltransferase 1A1 enzyme, UGT1A3 – uridine diphosphate glucuronosyltransferase 1A3 enzyme, UGT1A7 – uridine diphosphate glucuronosyltransferase 1A7 enzyme, UGT1A8 – uridine diphosphate glucuronosyltransferase 1A8 enzyme, UGT1A9 – uridine diphosphate glucuronosyltransferase 1A9 enzyme, UGT1A10 – uridine diphosphate glucuronosyltransferase 1A10 enzyme
The enzymes belonging to the group of human cytochrome P450 enzymes are responsible for the production of cholesterol, prostacyclin, and thromboxane A2, among other products. They participate in the biotransformation of drugs in phase 1 biotransformation [66, 67]. Cytochrome P450 enzymes are expressed primarily in the liver but also in the small intestine, lungs, and kidneys [66]. In humans, there are over 57 types of CYPs, which are divided into 18 families and 43 subfamilies [62]. Cytochrome P450 isoforms belonging to the CYP1, CYP2, and CYP3 families are responsible for the metabolism of approximately 80% of drugs [68].
On the other hand, UGT enzymes participate in the second phase of drug biotransformation [67] and are also believed to be involved in intracellular processes such as apoptosis. UGTs are found not only in the liver but also in the small intestine, bladder, and kidneys. This group is divided into two families (UGT1 and UGT2) and three subfamilies (UGT1A, UGT2A, and UGT2B) [69].
Many studies have demonstrated the impact of food on the activity of drug-metabolizing enzymes [70–73]. Food products, specifically the chemical compounds contained within them, act as inducers, inhibitors, or substrates for individual enzymes, thereby increasing or decreasing the concentration of a given drug in the plasma, which affects the treatment outcome and toxicity [43]. Table 4 shows the impact of popular dietary supplements on the metabolism of TKIs used in advanced RCC.
Table 4
The impact of popular dietary supplements on the metabolism of tyrosine kinase inhibitors used in advanced renal cell carcinoma [43, 45, 74, 75]
During metabolism, blueberry (Vaccinium corymbosum), elderberry (Sambucus nigra), evening primrose (Oenothera biennis), fenugreek (Trigonella foenum-graecum), garlic (Allium sativum), lavender (Lavandula angustifolia), olive (Olea europaea), papaya (Carica papaya), and valerian (Valeriana officinalis) do not interact with cabozantinib, lenvatinib, or sunitinib. There is a lack of information in the literature regarding the interactions during the metabolism of axitinib, pazopanib, sorafenib, and tivozanib with the aforementioned food products [75].
In the literature, there is a lack of data regarding interactions with TKIs used in RCC at the metabolism stage of the following food products: bladder wrack (Fucus vesiculosus), flaxseed (Linum usitatissimum), maté (Ilex paraguariensis), shiitake (Lentinula edodes), alfalfa (Medicago sativa), lemon balm (Melissa officinalis), guarana (Paullinia cupana), kombucha (Medusomyces gisevii), oyster mushroom (Pleurotus ostreatus), sorrel (Rumex acetosella), salvia (Salvia divinorum), chia (Salvia hispanica), wheat grass (Triticum aestivum), tea tree oil (Melaleuca alternifolia), bilberry (Vaccinium myrtillus), manuka honey (honey produced from the nectar of the Manuka tree – Leptospermum scoparium), psyllium (Plantago ovata), royal jelly (a secretion produced by worker bees – Apis mellifera), shark cartilage (cartilage derived from sharks – class Chondrichthyes).
An interesting issue is the role of the gut microbiome in drug metabolism. This activity may result in a different metabolite profile than that produced by host cells. It may be associated with varying levels of activation or inactivation of the pharmacological and/or toxic effects of a drug compared with those metabolites that are the result of transformations in host cells [45]. Research is ongoing to assess whether probiotic supplementation can influence the treatment effects of, for example, TKIs in advanced kidney cancer [76].
Once ethanol is introduced into the body, it undergoes all pharmacodynamic and pharmacokinetic transformations similar to those of a drug [41]. It also influences the course of these processes in the case of TKIs. In the presence of ethanol, the solubility of the TKIs used to treat advanced RCC increases, which increases the gradient between the intestine and plasma, consequently increasing the amount of drug in the plasma. This may lead to an increase in drug toxicity [45]. Ethanol is metabolized by alcohol dehydrogenase and CYP2E1 [68]. In this context, there is no competition between TKIs and ethanol [21–27]. This specifically pertains to pure ethanol.
Excretion
The last stage of the pharmacokinetic phase is excretion. This process involves elimination through bile and urine [61, 77]. For a substance to be excreted, it must be polar [41]. The drug-food interaction at this stage is related to the effect of consumed food on the speed of this process [61]. Tyrosine kinase inhibitors in RCC are primarily excreted in the feces, followed by the urine [21–27]. High-fat meals stimulate bile secretion, which may lead to a decrease in the bioavailability of these drugs [61]. Tyrosine kinase inhibitors belong to the group of weak bases [28]. The consumption of animal-derived foods such as fish, meat, eggs, and cheeses lowers the pH of the urine, thereby accelerating the excretion of these drugs through the kidneys [41].
Conclusions
Tyrosine kinase inhibitors represent a cornerstone in the treatment of advanced or metastatic RCC. A significant advantage of these agents is their oral administration, which enables patients to undergo therapy in the comfort of their own homes. However, the concomitant intake of these medications can lead to interactions, not only with other drugs but also with certain foods. Such interactions may impact both the tolerability and efficacy of the treatment. Therefore, understanding potential TKI-food interactions is crucial for optimizing the safety and therapeutic outcomes in patients with advanced RCC.






