
Summary
Tumor necrosis factor-like weak inducer of apoptosis (TWEAK), the level of which is high on the first day after myocardial infarction (MI), plays a key role in the development of adverse left ventricular remodeling. Higher levels of TWEAK at 1 day post-MI can induce matrix metalloproteinases. Increased sCD163 levels at 2 weeks post-MI seem to be associated with the healing process through neutralizing the excessive inflammatory effects of TWEAK.
Introduction
Cardiac remodeling is of primary importance for morbidity and mortality following acute myocardial infarction (MI) [1]. Post-MI cardiac remodeling includes a series of complex interplays, initiated by tissue damage and necrosis with subsequent sterile inflammation and infiltration by immune cells including neutrophils, monocytes/macrophages, dendritic cells, and lymphocytes [2], followed by the resolution of this inflammation characterized by myofibroblast proliferation, scar formation, and neovascularization [3]. Although inflammation is an important component of tissue healing, an excess inflammatory response contributes to adverse remodeling (AR) [4].
Proinflammatory cytokine levels are also elevated during the post-MI period. Tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) is a member of the TNF superfamily that plays a role in various biological activities including proliferation, migration, differentiation, apoptosis, angiogenesis, and inflammation. These biological activities are all closely related to the pathogenesis of cardiac remodeling. TWEAK carries out these activities by affecting endothelia, smooth muscle cells, cardiomyocyte cell lines, and cardiac fibroblasts [5–7]. Soluble CD163 (CD163) was discovered as the second receptor of TWEAK in 2007 [8]. CD163 is expressed by monocytes/macrophages and acts as a scavenger receptor, preventing its harmful biological outcomes [9]. CD163 interacts with TWEAK in vivo for the regulation of tissue regeneration following ischemic damage [10]. On the other hand, matrix metalloproteinases (MMPs), which play a role in tissue remodeling and degradation of extracellular matrix proteins, may be associated with adverse remodeling [11]. However, the roles of TWEAK/CD163 interaction and MMPs in cardiovascular pathology are still unclear.
Aim
In the present study, we aimed to investigate the roles of TWEAK, sCD163, and MMP in left ventricular (LV) AR in the early post-MI period.
Material and methods
Study design and population
This multi-centered prospective study was conducted between June 2015 and June 2018, and designed in compliance with the Helsinki Declaration, revised in Brazil in 2013, and Good Clinical Practice Guidelines. Ethics committee approval was obtained from the local ethics committee (Decision no: 2013/106). Informed consent forms of all of the patients enrolled in the study were also obtained. Assuming an α of 0.05, a power of 0.80, and a 30% estimated AR rate, in line with previous reports, the estimated sample size was at least 46 patients in total.
Forty-six patients who were admitted to the emergency department with first-time ST-elevation myocardial infarction (STEMI), who were above 18 years of age, presented within the first 12 h after the onset of a complaint of chest pain that had been ongoing for more than 30 min with > 0.1 mV ST-segment elevation in at least 2 or more related derivations and underwent primary percutaneous coronary intervention (PCI) were enrolled in the study. The definition of STEMI was based on the third universal definition of MI [12] and was managed according to the latest European Society of Cardiology (ESC) guidelines [13].
Patients in cardiogenic shock or in need of intra-aortic balloon pump, with a history of silent ischemia/infarct, any kind of systemic inflammatory disease, autoimmune disease, history of chronic corticosteroid or anti-inflammatory drugs, pregnancy or delivery within the previous 90 days or breastfeeding mothers, and patients for whom emergency or elective coronary artery bypass grafting was planned after angiography, and patients who had myocardial re-infarction during follow-up were excluded from the study. Clinical, demographic, laboratory, and radiological findings of patients were assessed.
After inclusion, follow-up cardiac magnetic resonance (CMR) scanning was performed at 2 weeks and 6 months after the index event. The assessment of inflammatory markers was conducted on the first day and 2 weeks (14 days) after the index event. The definition of AR was made considering the 10% change (∆) in the LV end-diastolic volume (LVEDV) at 6 months compared to 2 weeks (14 days) post-MI, which has been widely used in the literature [14].
Biochemical parameters
Blood samples were obtained from the antecubital vein upon admission for the troponin and lipid panel, and at presentation, at 1 day and 2 weeks post-MI for inflammatory markers. Samples were centrifuged at 1500 rpm for 10 min and stored at –80°C. The tests were analyzed after all of the samples were collected and run in the same laboratory by the same laboratory technician in a single session with the same device. The total cholesterol was determined using the homogeneous enzymatic colorimetric method in a Hitachi Modular P800 autoanalyzer (Roche Diagnostics Corp., Indiana, IN, USA). Low density lipoprotein (LDL) was calculated according to the Friedewald method [15].
Analysis of cytokine parameters
Cytokine quantification was run in duplicate. Serum and plasma samples were thawed on ice and concentrations of TWEAK, sCD163, MMP-1, MMP-2, and MMP-3 were analyzed according to the manufacturer’s instructions for the bead-based multiplex immunoassay system (171-AL001M, Bio-Plex Pro Human Inflammation Panel 1, 37-Plex). The formation of different sandwich immunocomplexes on distinct bead sets was measured and quantified using the Bio-Plex MAGPIX System (Bio-Rad Laboratories, Hercules, CA, USA). The final concentration of analytes was calculated using Bio-Plex Manager v5.0 software (Bio-Rad). For all of the statistical analyses, values below the detection limit of the assay were replaced with the minimal detectable value for the analyte. The coefficient of variation (CV%) was < 10%.
Cardiac magnetic resonance imaging
All of the CMR studies were performed on a 3-T scanner (Magnetom Skyra, Siemens Medical Systems, Erlangen, Germany). The cardiac MR imaging protocol included the acquisition of one 4-chamber view, cine short-axis sections (slice thickness 6 mm at 10-mm intervals) and one 2-chamber view. The indices of LV systolic function were assessed using a retrospective electrocardiogram-gated turbo-fast low angle shot (turbo-FLASH) sequence. The imaging parameters included an echo time of 1.42 ms, repetition time of 39 ms, flip angle of 57°, and voxel size of 1.67 × 1.67 × 6 mm. The LV function and volumes were measured using Syngo via imaging software. The LVEDV and LVESV were calculated with short-axis based planimetry from the basal to apical level. The stroke volume was calculated as the LVEDV minus the LVESV, and the LV ejection fraction (LVEF) was calculated as follows: LVEF = [(LVEDV – LVESV)/LVEDV] × 100.
Statistical analysis
The statistical analyses were performed using the IBM SPSS Statistics for Windows 20.0 (IBM Corp., Armonk, NY, USA) program. Normal distribution of data was evaluated using the Shapiro-Wilk test. Numeric variables with and without normal distribution were plotted as the mean ± standard deviation and median (interquartile range (IQR); 25th (Q1)–75th (Q3) quartiles) respectively. Categorical variables were indicated as numbers and percentages. Student’s t test or the Mann-Whitney U test was used for comparison of the numeric variables between the 2 groups according to the distribution of normality. The χ2, Yates correction, and Fisher exact tests were used for comparison of the categorical data. The relationship between the TWEAK and MMPs was examined by Spearman’s correlation test. Mixed model for repeated measures (MMRM) analysis was performed for comparison of the post-MI cytokine levels. Changes in CMR variables and cytokine levels in the post-MI period are shown by Δ. Potential risk factors for AR were defined by univariable logistic regression analysis. Variables with a p value less than 0.25 were included in the multivariable logistic regression models [16, 17]. Additionally, confounder factors, including age and sex, smoke, comorbidities, symptom to balloon time, infarct location, cardiac output and infarct size, were included in the multivariable regression models, because these parameters may play a role in LV function [18–20]. In multivariable regression Model I, confounding and potential factors and cytokine levels on the first day post-MI were included. In multivariable regression Model II, confounding and potential factors and change of cytokine levels from the first day to 2 weeks post-MI were included. P < 0.05 was accepted as statistically significant.
Results
The study population consisted of 6 women and 40 men (mean age: 55.5 ±7.1 years). Seventeen patients (37%) had hypertension and the rate of diabetes was 26.1% (n = 12). Eighteen (39.1%) patients exhibited AR post-MI. Details of demographic, laboratory and CMR imaging findings are shown in Table I.
Table I
Demographic, laboratory, and cardiac magnetic resonance imaging baseline findings
| Variables | All population N = 46 | Adverse remodeling | P-value | |
|---|---|---|---|---|
| No n = 28 | Yes n = 18 | |||
| Demographic findings: | ||||
| Age [years] | 55.5 ±6.9 | 55.8 ±7.2 | 54.9 ±6.4 | 0.658 |
| Male gender, n (%) | 40 (87.0) | 25 (89.3) | 15 (83.3) | 0.891 |
| BMI [kg/m2] | 27.1 ±4.0 | 26.8 ±4.3 | 27.5 ±3.4 | 0.565 |
| Smoking, n (%) | 18 (39.1) | 9 (32.1) | 9 (50.0) | 0.354 |
| Hypertension, n (%) | 17 (37.0) | 10 (35.7) | 7 (38.9) | 0.999 |
| Diabetes mellitus, n (%) | 12 (26.1) | 7 (25.0) | 5 (27.8) | 0.999 |
| SBP [mm Hg] | 125.0 ±15.8 | 124.5 ±16.4 | 125.8 ±15.2 | 0.786 |
| DBP [mm Hg] | 78.9 ±12.3 | 78.8 ±13.2 | 79.1 ±11.1 | 0.939 |
| Heart rate [bpm] | 82.2 ±10.1 | 81.4 ±11.5 | 83.3 ±8.0 | 0.509 |
| Door-to-balloon time [min] | 48.1 ±6.8 | 47.8 ±7.5 | 48.6 ±5.7 | 0.693 |
| Symptom-to-balloon time [min] | 350.8 ±70.4 | 352.8 ±71.9 | 347.8 ±70.1 | 0.818 |
| Anterior infarct, n (%) | 27 (58.7) | 16 (57.1) | 11 (61.1) | 0.999 |
| Laboratory findings: | ||||
| cTn-I [pg/ml] | 47.8 (30.4–58.6) | 40.4 (30.4–52.6) | 57.3 (40–67.9) | 0.033* |
| WBC [× 103 µl] | 11.4 ±2.6 | 11.2 ±2.5 | 11.7 ±2.9 | 0.490 |
| Neutrophils [× 103 µl] | 8.6 ±2.2 | 8.1 ±2.2 | 9.6 ±2.1 | 0.027* |
| Lymphocytes [× 103 µl] | 2.3 ±0.7 | 2.6 ±0.7 | 2.1 ±0.6 | 0.016* |
| Platelets [× 103 µl] | 239.5 ±41.7 | 238.3 ±47.2 | 241.5 ±32.8 | 0.800 |
| Monocytes [× 103 µl] | 0.8 ±0.2 | 0.8 ±0.2 | 0.9 ±0.2 | 0.105 |
| Hemoglobin [g/dl] | 13.7 ±1.4 | 13.7 ±1.3 | 13.8 ±1.6 | 0.765 |
| Glucose [mg/dl] | 115 (96–130) | 115.5 (96–136.5) | 113 (90–130) | 0.479 |
| Creatinine [mg/dl] | 1.0 ±0.2 | 1.0 ±0.3 | 1.0 ±0.2 | 0.779 |
| hs-CRP [µg/l] | 21.6 (14.2–31) | 19.1 (12.8–27.9) | 23.9 (14.2–34.5) | 0.180 |
| Total cholesterol [mg/dl] | 179.2 ±51.6 | 175.4 ±56 | 185.2 ±44.9 | 0.534 |
| LDL [mg/dl] | 111 (90–137) | 111 (59.5–134) | 112 (96–145) | 0.150 |
| HDL [mg/dl] | 38.5 ±8.2 | 36.9 ±5.2 | 41.1 ±11.1 | 0.312 |
| Triglyceride [mg/dl] | 120.5 (72–204) | 124.5 (70–215.5) | 116.5 (90–146) | 0.890 |
| CMR imaging findings: | ||||
| 2 weeks post-MI: | ||||
| LVEF (%) | 47.0 ±10.5 | 45.9 ±10.2 | 48.8 ±10.9 | 0.200 |
| LVEDV [ml] | 160.4 ±33.0 | 165.9 ±35.4 | 153.5 ±24.4 | 0.517 |
| LVESV [ml] | 89 (61–140) | 88.5 (61–140) | 91 (56–118) | 0.418 |
| Stroke volume [ml] | 72.6 ±13.2 | 69.8 ±9.7 | 76.9 ±16.8 | 0.111 |
| CO [ml/min] | 4.5 ±1.0 | 4.3 ±0.8 | 4.8 ±1.3 | 0.137 |
| CI [ml/min/m2] | 2.5 ±0.4 | 2.4 ±0.4 | 2.6 ±0.4 | 0.113 |
| Infarct size (%) of LV | 17 (9–26) | 14 (8–22) | 18 (12–26) | 0.042* |
| 6 months post-MI: | ||||
| LV EF (%) | 48.5 ±10.4 | 50.8 ±8.7 | 44.4 ±11.0 | 0.034* |
| LV EDV [ml] | 159.8 ±34.3 | 146.1 ±30.5 | 181.0 ±29.3 | < 0.001* |
| LV ESV [ml] | 80 (62–131) | 74.5 (53–124) | 106 (70–153) | 0.015* |
| Stroke volume [ml] | 75.0 ±14.6 | 75.7 ±12.2 | 74.0 ±18.1 | 0.701 |
| Cardiac output [ml/min] | 4.6 ±1.2 | 4.7 ±0.9 | 4.5 ±1.6 | 0.616 |
| Cardiac index [ml/min/m2] | 2.6 ±0.4 | 2.7 ±0.4 | 2.5 ±0.5 | 0.387 |
| Infarct size (%) of LV | 15 (6–20) | 11 (4–18) | 16 (10–23) | 0.020* |
Numerical variables are shown as mean ± standard deviation or median (IQR). Categorical variables are shown as numbers (%).
AR – adverse remodeling, CI – cardiac index, CMR – cardiac magnetic resonance, CO – cardiac output, cTn-I – cardiac troponin I, DBP – diastolic blood pressure, LDL – low density lipoprotein, LVEF – left ventricular ejection fraction, LVEDV – left ventricular end-diastolic volume, LVESV – left ventricular end-systolic volume, SBP – systolic blood pressure.
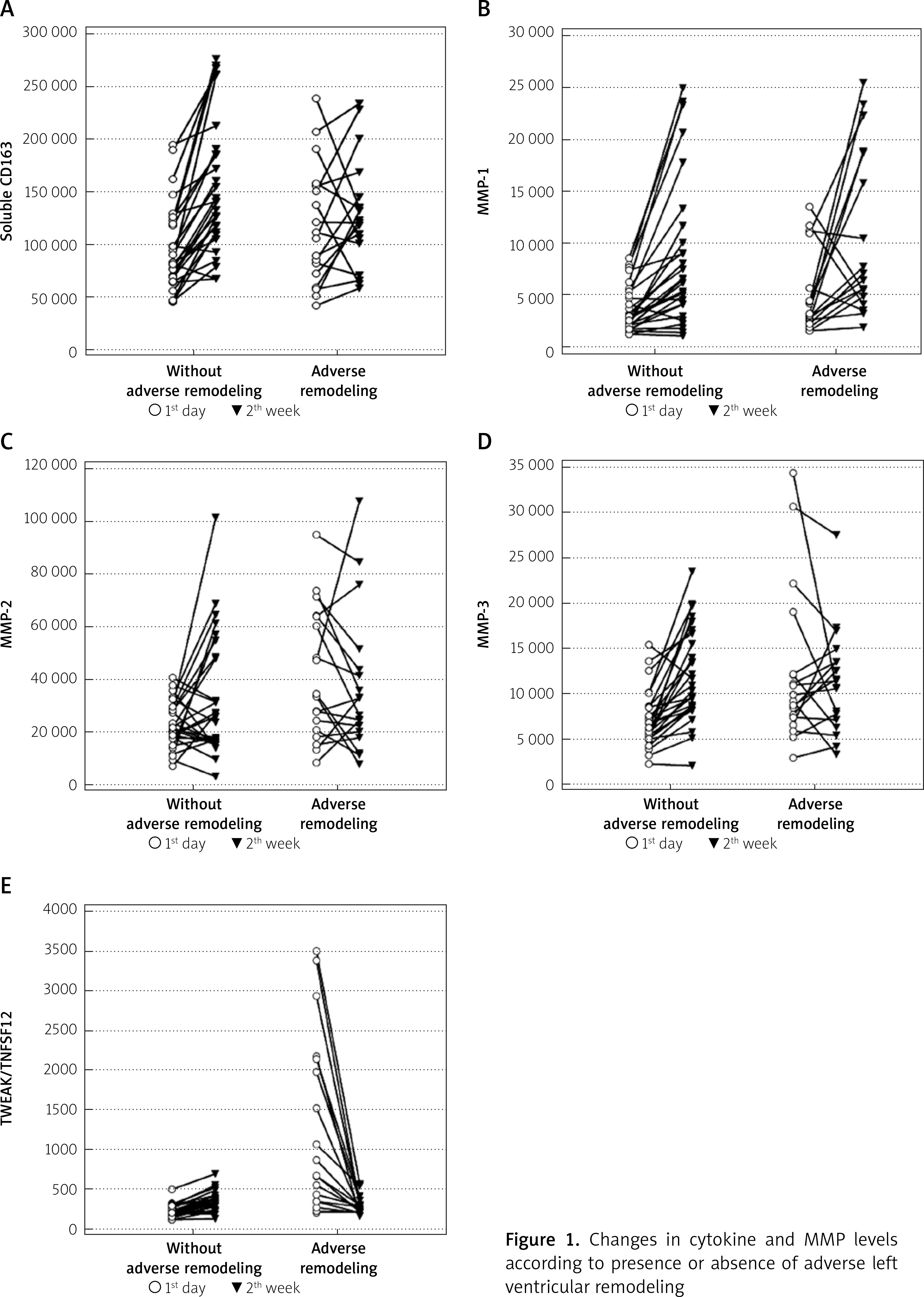
TWEAK, MMP-2 and MMP-3 levels on the first day post-MI were higher in the patients with AR. The cytokine levels increased at 2 weeks compared to the first day post-MI in patients without AR. Median MMP-1 levels was increased at 2 weeks compared to the first day post-MI in patients with AR (3072.5 vs. 6695.6 pq/ml, p = 0.016), TWEAK level was decreased (759.4 vs. 276.5 pq/ml, p < 0.001), and other cytokine levels showed no significant change (Figure 1, Table II).
Table II
Changes in cytokine and MMP levels in the post-MI period
| Variables | Adverse remodeling | 1st day | P-value1 | 2nd week | P-value2 | P-value3 | Δp | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Median | Q1 | Q3 | Median | Q1 | Q3 | ||||||
| TWEAK [pg/ml] | No, n = 28 | 216.0 | 168.7 | 242.8 | < 0.001* | 336.4 | 250.0 | 375.6 | 0.074 | < 0.001* | < 0.001* |
| Yes, n = 18 | 759.4 | 342.9 | 2134.0 | 276.5 | 208.4 | 297.6 | < 0.001* | ||||
| sCD163 [pg/ml] | No, n = 28 | 82675.6 | 68811.1 | 120855.0 | 0.242 | 139870.2 | 109724.9 | 187761.1 | 0.192 | <0.001* | 0.008* |
| Yes, n = 18 | 108351.9 | 71936.7 | 154971.8 | 121487.3 | 100603.4 | 144322.7 | 0.327 | ||||
| MMP-1 [pg/ml] | No, n = 28 | 2799.9 | 1919.6 | 5092.4 | 0.265 | 6469.0 | 4055.0 | 10835.1 | 0.507 | < 0.001* | 0.838 |
| Yes, n = 18 | 3072.5 | 2446.6 | 5597.7 | 6695.6 | 4833.7 | 18667.2 | 0.016* | ||||
| MMP-2 [pg/ml] | No, n = 28 | 20987.5 | 17506.3 | 28579.7 | 0.010* | 25572.8 | 16294.2 | 48138.4 | 0.471 | 0.045* | 0.043* |
| Yes, n = 18 | 29316.2 | 20728.2 | 63681.1 | 30526.1 | 19906.7 | 48138.4 | 0.231 | ||||
| MMP-3 [pg/ml] | No, n = 28 | 6156.5 | 4978.1 | 8472.8 | 0.005* | 10563.1 | 8425.4 | 15980.4 | 0.500 | < 0.001* | 0.001* |
| Yes, n = 18 | 9493.1 | 7364.4 | 12069.3 | 10984.1 | 7031.5 | 13423.5 | 0.845 | ||||
P-value1: Comparison of 1st day results (adverse left ventricular remodeling groups: No vs. Yes). P-value2: Comparison of 2nd week results (adverse left ventricular remodeling groups: No vs. Yes). P-value3 = 1st day vs. 6th week in groups with and without adverse left ventricular remodeling. Δp = Comparison of the changes of follow-ups between the adverse left ventricular remodeling groups (No vs. Yes). TWEAK – tumor necrosis factor like weak inducer of apoptosis, MMP – matrix metalloproteinases.
Figure 1
Changes in cytokine and MMP levels according to presence or absence of adverse left ventricular remodeling
There was a positive correlation between the TWEAK and MMP levels post-MI (Table III). Potential risk factors of AR were determined by univariable logistic regression analysis (Table IV). Multivariable regression Model I indicated that risk factors of high TWEAK and high MMP-3 levels on the first day post-MI were independent predictors of AR (OR = 1.03, 95% CI: 1.01–1.05, p = 0.006; OR = 1.08, 95% CI: 1.04–1.13, p = 0.015; respectively). Accordingly; on the first day post-MI, an increase of 1 pq/ml in TWEAK levels increased the risk of developing AR 1.03-fold; and an increase of 1 pq/ml in MMP-3 levels increased the risk of developing AR 1.08-fold (Table IV).
Table III
Relationship between TWEAK, sCD163 and MMPs in the post-MI period
| Variables | Correlation | 1st day n = 46 | 2nd week n = 46 | Change (2th week–1st day) n = 46 | |||
|---|---|---|---|---|---|---|---|
| TWEAK | sCD163 | TWEAK | sCD163 | TWEAK | sCD163 | ||
| sCD163 | r | 0.445 | – | 0.711 | – | 0.591 | – |
| P-value | 0.002* | – | < 0.001* | – | < 0.001* | – | |
| MMP-1 | r | 0.338 | 0.399 | 0.378 | 0.604 | 0.329 | 0.388 |
| P-value | 0.022* | 0.006* | 0.010* | < 0.001* | 0.031* | 0.008* | |
| MMP-2 | r | 0.425 | 0.361 | 0.493 | 0.402 | 0.416 | 0.347 |
| P-value | 0.001* | 0.014* | 0.001* | 0.006* | 0.004* | 0.014* | |
| MMP-3 | r | 0.479 | 0.225 | 0.590 | 0.516 | 0.611 | 0.619 |
| P-value | 0.001* | 0.134 | < 0.001* | < 0.001* | < 0.001* | < 0.001* | |
Table IV
Effect of cytokines and MMPs on adverse left ventricular remodeling
| Variables | Univariable | Model I multivariable | Model II multivariable | ||||||
|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | P-value | OR | 95% CI | P-value | OR | 95% CI | P-value | |
| Demographic parameters: | |||||||||
| Age [years] | 0.98 | 0.90–1.07 | 0.650 | – | – | – | – | – | – |
| Male gender | 0.60 | 0.11–3.36 | 0.887 | – | – | – | – | – | – |
| BMI | 1.05 | 0.90–1.22 | 0.557 | – | – | – | – | – | – |
| Smoking | 2.11 | 0.63–7.13 | 0.290 | – | – | – | – | – | – |
| Hypertension | 1.15 | 0.34–3.90 | 0.950 | – | – | – | – | – | – |
| Diabetes mellitus | 1.15 | 0.30–4.41 | 0.946 | – | – | – | – | – | – |
| SBP | 1.01 | 0.97–1.04 | 0.780 | – | – | – | – | – | – |
| DBP | 1.01 | 0.95–1.05 | 0.937 | – | – | – | – | – | – |
| Heart rate | 1.02 | 0.96–1.08 | 0.500 | – | – | – | – | – | – |
| Door-to-balloon time | 1.02 | 0.93–1.11 | 0.685 | – | – | – | – | – | – |
| Symptom-to-balloon time | 0.99 | 0.90–1.01 | 0.813 | – | – | – | – | – | – |
| Anterior infarct | 1.18 | 0.35–3.94 | 0.963 | – | – | – | – | – | – |
| Laboratory parameters: | |||||||||
| cTn-I | 1.10 | 1.01–1.20 | 0.030* | – | – | – | – | – | – |
| WBC | 1.08 | 0.86–1.37 | 0.482 | – | – | – | – | – | – |
| Neutrophil | 1.19 | 1.03–1.36 | 0.024* | – | – | – | – | – | – |
| Lymphocyte | 0.37 | 0.14–0.61 | 0.015* | – | – | – | – | – | – |
| Platelet | 1.01 | 0.98–1.03 | 0.795 | – | – | – | – | – | – |
| Monocyte | 1.01 | 0.98–1.06 | 0.100 | – | – | – | – | – | – |
| Hemoglobin | 1.07 | 0.69–1.66 | 0.758 | – | – | – | – | – | – |
| Glucose | 0.99 | 0.98–1.02 | 0.473 | – | – | – | – | – | – |
| Creatinine | 1.49 | 0.10–22.60 | 0.773 | – | – | – | – | – | – |
| hs-CRP | 1.03 | 0.97–1.08 | 0.172 | – | – | – | – | – | – |
| Total cholesterol | 1.01 | 0.99–1.02 | 0.526 | – | – | – | – | – | – |
| LDL | 1.07 | 0.99–1.20 | 0.147 | – | – | – | – | – | – |
| HDL | 1.01 | 0.98–1.03 | 0.301 | – | – | – | – | – | – |
| Triglyceride | 0.99 | 0.95–1.11 | 0.872 | – | – | – | – | – | – |
| CMR imaging parameters: | – | – | – | ||||||
| LVEF | 1.03 | 0.97–1.09 | 0.184 | – | – | – | – | – | – |
| Stroke volume | 1.05 | 0.98–1.10 | 0.084 | – | – | – | – | – | – |
| CO | 1.59 | 0.85–2.97 | 0.134 | – | – | – | – | – | – |
| CI | 3.47 | 0.72–16.74 | 0.101 | – | – | – | – | – | – |
| Infarct size | 1.08 | 1.02–1.13 | 0.040* | – | – | – | – | – | – |
| Cytokines: | |||||||||
| 1st day post-MI: | |||||||||
| TWEAK | 1.02 | 1.01–1.03 | < 0.001* | 1.03 | 1.01–1.05 | 0.006* | – | – | – |
| sCD163 | 1.01 | 0.98–1.05 | 0.154 | – | – | – | – | – | – |
| MMP1 | 1.01 | 0.99–1.04 | 0.148 | – | – | – | – | – | – |
| MMP2 | 1.05 | 1.01–1.10 | 0.024* | – | – | – | – | – | – |
| MMP3 | 1.07 | 1.02–1.12 | 0.007* | 1.08 | 1.04–1.13 | 0.015* | – | – | – |
| Nagelkerke R2 = 0.54; p < 0.001* | |||||||||
| Change: | – | – | – | ||||||
| ΔTWEAK | 0.90 | 0.82–0.97 | 0.015* | – | – | – | 0.92 | 0.84–0.99 | 0.018* |
| ΔsCD163 | 0.98 | 0.98–0.99 | 0.037* | – | – | – | – | – | – |
| ΔMMP1 | 1.00 | 0.99–1.01 | 0.310 | – | – | – | – | – | – |
| ΔMMP2 | 0.97 | 0.95–0.99 | 0.049* | – | – | – | – | – | – |
| ΔMMP3 | 0.96 | 0.94–0.98 | 0.005* | – | – | – | 0.98 | 0.96–0.99 | 0.005* |
| Nagelkerke R2 = 0.46; p < 0.001* | |||||||||
Model II regression analysis indicated that the changes of TWEAK and MMP-3 levels from the first day to 2 weeks post-MI were independent predictors of AR (OR = 0.92, 95% CI: 0.84–0.99, p = 0.018; OR = 0.98, 95% CI: 0.96–0.99, p = 0.005; respectively). Accordingly, a decrease of 1 pq/ml in ΔTWEAK levels increased the risk of developing AR 1.08 (1/0.92)-fold, and an decrease of 1 pq/ml in ΔMMP-3 levels increased the risk of developing AR 1.02 (1/0.98)-fold (Table IV).
Discussion
The first day after MI, TWEAK, MMP-2, and MMP-3 levels were higher in the patients with AR, while there was no significant difference between the groups with regard to sCD163. The cytokine levels between groups with AR and without AR at 2 weeks post-MI were not significantly different. The TWEAK expression decreased at 2 weeks post-MI in the patients with AR, while no significant change was observed in the sCD163, MMP-2, and MMP-3 expression levels. The cytokine levels in the patients without AR increased significantly at 2 weeks post-MI compared to the baseline levels on first day post-MI. Regression analyses indicated that the higher levels of TWEAK and MMP-3 on the first day post-MI contributed to the development of AR.
The process of cardiac remodeling includes structural and functional changes, such as cardiomyocyte proliferation, hypertrophy, necrosis, apoptosis, autophagy, interstitial fibrosis, contractile dysfunction, and ventricular dilatation [21]. Several experimental studies have revealed the upregulation of TWEAK in cardiomyocytes after experimental MI [22, 23]. A study by Jarr et al. [24] on rats indicated that TWEAK caused post-MI negative effects on cardiomyocytes in terms of cardiac repair by inhibiting peroxisome proliferator activated receptor γ co-activator 1α, thereby decreasing metabolic adaptation and increasing cardiac workload. Jain et al. [22] discovered that increased TWEAK levels were associated with severe cardiac dysfunction in a study conducted with mouse and human cardiomyocytes. A study by Unudurthi et al. [25] showed that TWEAK caused cardiac fibrosis by triggering macrophage infiltration in rats. Another study on mice reported that TWEAK caused progressive dilated cardiomyopathy [26]. The inhibition of peroxisome proliferator activated receptor gamma co-activator 1α by TWEAK was the mechanism in the development of dilated cardiomyopathy [26]. Similar to the above-mentioned study, the current study revealed that the TWEAK level increase that was observed after MI was associated with cardiac AR.
Membrane protein CD163 is one of the TWEAK receptors. CD163 is a scavenger receptor expressed by monocytes and macrophages, scavenging hemoglobin-haptoglobin (Hb-Hp) complexes and protecting tissues from oxidative damage and inflammation. TWEAK has affinity to the CD163 receptor due to the molecular similarity between the Hb-Hp complex and TWEAK [27]. The combination of TWEAK and CD163 causes the neutralization of both molecules. In this respect, CD163 is also known as a TWEAK decoy receptor [8]. Several studies have reported an increase in CD163 levels due to increased TWEAK levels [28, 29]. This increase occurs in order to both neutralize the negative effects of TWEAK and initiate its effects via the nuclear factor-κB signaling pathway. In accordance with these findings, a significant positive correlation was found herein between the TWEAK levels and CD163 at first day and 2 weeks post-MI. In addition, there was a significant positive correlation between the temporal changes in the TWEAK and CD163 levels. Furthermore, the CD163 levels reached statistical significance in the patients without AR. These results suggested that CD163 caused the enhanced neutralization of the negative effects of TWEAK in the patients without AR, resulting in improved cardiac repair.
MMPs are members of the zinc-containing endoprotease family responsible for tissue remodeling and the degradation of extracellular matrix proteins, released by endothelia, vascular smooth muscle cells, fibroblasts, osteoblasts, macrophage neutrophils, and lymphocytes. MMP levels increase shortly after MI, leading to an imbalance between the MMPs and tissue inhibitors of MMPs in cardiac tissue. This imbalance causes degradation of the cardiac extracellular matrix. Cardiac tissue thickness decreases after degradation, causing impaired cardiac structure and function [30, 31]. Wegiel and Rakowski [32] examined the relationship between biomarkers and remodeling in 53 articles between 2005 and 2020. The authors found that 160 biomarkers, a significant portion of which are MMPs, are associated with cardiac remodeling, and a positive correlation was found between MMP-3 and cardiac remodeling in 3 of the 4 studies reviewed. Pecherina et al. [33] reported a positive relation between MMP-3 levels and diastolic dysfunction, LVEF, and LVESV. Fan et al. [34] conducted a preclinical study and postulated that MMP-2 causes AR in the post-MI period as a result of extracellular matrix damage. They developed a MMP-2 inhibitor and injected it to infarcted heart tissue for 4 weeks post-MI. They discovered that this inhibitor not only increases tissue thickness, but also effectively prevents cardiac extracellular matrix degradation by preserving the collagen composition of normal cardiac tissue. They reported significant improvement in cardiac function without the induction of fibrosis and showed that AR can be effectively prevented by MMP-2 inhibition. In the current study, it was discovered that the MMP-2 and MMP-3 levels were higher in the patients with AR than in those without AR on first day post-MI. The univariable regression analysis indicated that the elevated MMP-2 and MMP-3 levels on the first day post-MI contributed to the development of AR. In addition, the finding of a positive correlation between the TWEAK and MMP levels in the early post-MI period supported the concept that TWEAK can induce MMPs by activating the NF-κB pathway [6, 35, 36]. These results suggested that an excessive post-MI inflammatory response activates MMPs and these collagenolytic MMPs cause extracellular matrix damage and cardiac fibrosis, resulting in impairment of cardiac function and structure.
Cardiac remodeling can occur weeks to months after reperfusion. Increasing evidence indicates that TWEAK and MMP levels may be an important biomarker in detecting high-risk patients post-MI. Typical measurement methods may not fully reflect the in vivo activity of cytokines. In addition, ideal biomarkers should be easily detectable from blood [32]. Therefore, the routine use of cytokine measurements can be difficult. Furthermore, TWEAK and MMPs may be a new therapeutic target in adverse cardiac remodeling. In high-risk patients post-MI, ACE inhibitors and angiotensin II receptor inhibitors and statins may decrease concentrations of TWEAK and MMP-3 [37, 38].
The main limitation of this study was the small size of the study population. Another limitation was that the TWEAK level was a systemically measured parameter. Local cardiomyocyte levels of TWEAK should also be measured in order to determine whether TWEAK is effective in cardiac remodeling by acting on cardiomyocytes. This is not possible in human models. However, TWEAK upregulation has been reported in the cardiomyocytes of animal models [24].
Conclusions
There is a positive correlation between TWEAK and MMP-3 levels after MI, and excessive increases in both cytokine levels are associated with the development of AR. The results of the present study suggest that increased post-MI sCD163 expression may neutralize the excessive inflammatory effects of TWEAK.







