
Introduction
Liver diseases have continued to impose a heavy burden on the global healthcare system. Particularly, under the International Agency for Research on Cancer (IARC), it was estimated that there were 865,269 newly diagnosed liver cancer patients in 2022, accounting for 4.3% of the new cases of all cancers, making it the sixth most common cancer and contributing to 757,948 deaths, accounting for 7.8% of the total mortality rate among all cancers, ranking as the third most lethal cancers [1]. Currently, hepatocellular carcinoma (HCC) is the most common liver cancer and thus needs to be extensively studied. As precursor stages of liver cancer, the pathogenesis of liver fibrosis (LF) and cirrhosis warrants further exploration. LF is a dynamic process characterized by excessive accumulation of the extracellular matrix caused by hepatic stellate cell activation [2]. If untreated, LF can progress to cirrhosis or HCC, which increases the risk of death [3]. The incidence of LF has been on the rise globally, but there are no effective drugs for preventing or inhibiting it [4]. As the end stage of chronic liver disease, cirrhosis is the 11th leading cause of death globally [5], and is often associated with the development of LF [6], which, in turn, develops into liver cancer [7]. This calls for investigations into the development of novel therapeutic approaches for LF, cirrhosis, and HCC.
Notably, the development and testing of novel drugs is an expensive and time-consuming process with a high risk of failure [8]. To address the shortcomings of these traditional approaches of drug development, several research tools, including genomics, transcriptomics, proteomics, and other omics, have been designed in recent years, leading to the era of big data [9]. Using multi-omics data, researchers have been able to identify new drug targets for diseases, suggesting that multi-omics analyses shorten the development time and cost of developing new medicines.
In recent years, MR has increasingly been applied to study disease mechanisms using data from genome-wide association studies (GWAS). MR uses genetic variation data from GWAS to probe causal relationships between exposures and outcomes, and is considered to be similar to randomized controlled trials, presenting strong evidence because alleles are randomly assigned to the offspring at conception [10]. Recently, several researchers have investigated therapeutic targets for various diseases through genomic or proteomic analyses based on cis-expression quantitative trait locus (cis-eQTL) or cis-protein quantitative trait locus (cis-pQTL) data by MR [11–13]. In this study, we used genomic and proteomic analyses to identify the therapeutic targets of LF, cirrhosis, and HCC through MR. Subsequently, potential targets were identified through colocalization analysis. In addition, we conducted external validation for the identified targets using summary-data-based MR (SMR). To further provide important information to guide the development of effective drugs, we conducted a preliminary exploration of the regulatory mechanisms of the key targets.
Material and methods
Ethics statement
There were no ethical issues involved in our study as the data sources for this study were opensource databases, and ethical approvals for them have been obtained.
Study design
In this study, novel therapeutic targets for liver diseases (here, LF, cirrhosis, and HCC) were screened using dual-omics analysis at the gene and protein levels. An overview of this study is presented in Figure 1. Initially, we explored the causal association of druggable genes and proteins with liver diseases through a two-sample MR analysis based on cis-eQTL and cis-pQTL to identify potential therapeutic targets for these diseases. Subsequently, colocalization analyses were performed to determine whether the candidate drug targets shared genetic variation with liver diseases to select significant candidate targets. The SMR approach was employed to validate the causal relationships between significant candidate drug targets and liver diseases. Finally, a preliminary exploration of the regulatory mechanisms of the selected targets was conducted through a two-step MR, and the mediating roles of circulating metabolites, immune cells, gut microbiota, and inflammatory proteins were analyzed.
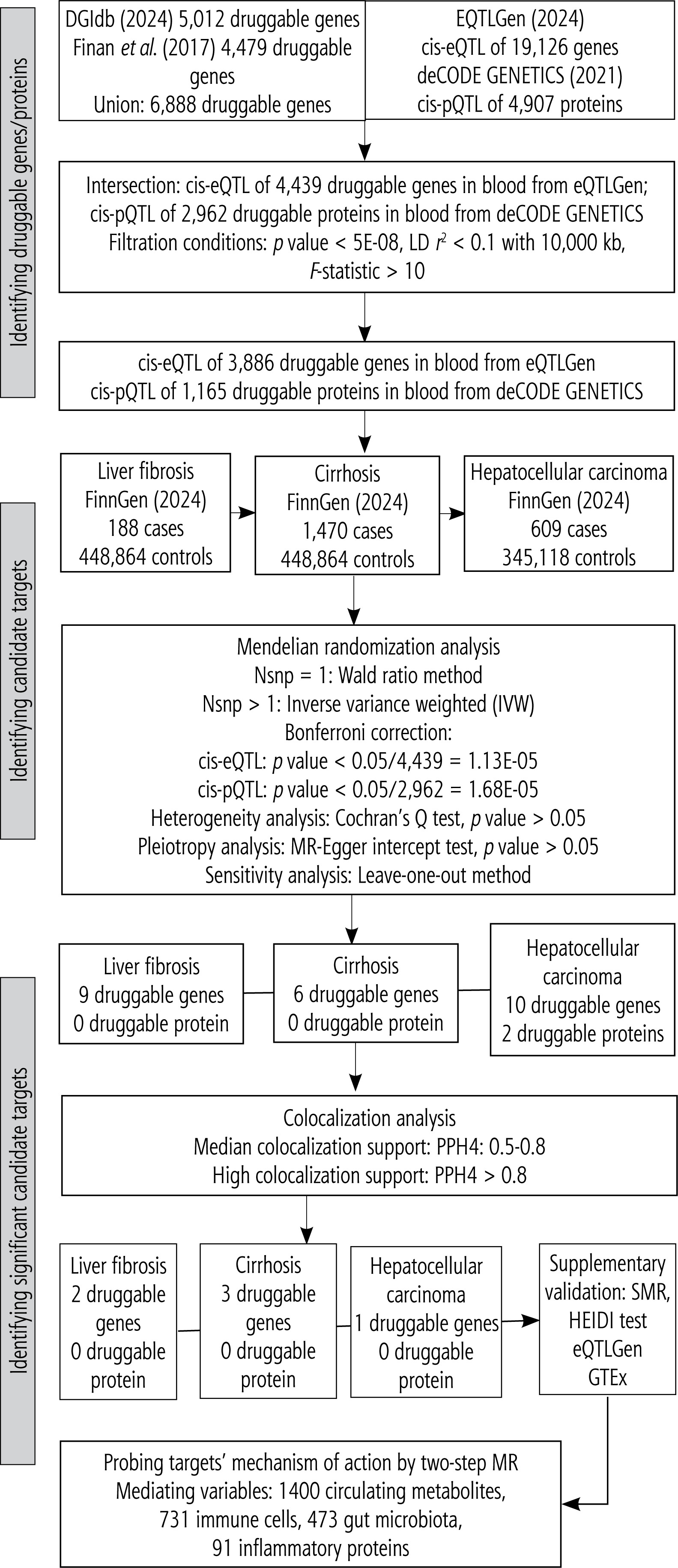
Exposure data
The druggable genes included in the MR analysis were obtained from the Drug-Gene Interaction Database (DGIdb, https://www.dgidb.org/) [14] and the study of Finan et al. [15]. DGIdb is a database containing drug-gene interactions and is widely used in genetics and pharmacology research [14]. The DGIdb was screened to download the latest “interactions” data updated in May 2024. A total of 5012 druggable genes were extracted from it. Moreover, 4479 druggable genes were extracted from the study by Finan et al. The total druggable genes obtained from both sources were merged, totaling 6888 druggable genes.
To identity the candidate therapeutic targets for the diseases at the gene level, MR analysis was performed based on the expression quantitative trait locus (eQTL). Generally, eQTL are divided into cis and trans, and the exposure data were the cis-eQTL data, representing genes normally located in the upstream and downstream 1 Mb regions of their regulatory genes. The updated eQTLGen Consortium (https://eqtlgen.org/) contained cis-eQTL data in blood samples for 19,126 genes. The intersection of these genes with 6,888 druggable genes yielded cis-eQTL data for 4,439 druggable genes, which served as the exposure data in this study.
To identify the candidate therapeutic targets for the selected diseases at the protein level, we selected pQTL as the exposure variable. The deCODE GENETICS (https://www.decode.com/) was screened to download the protein quantitative trait locus (pQTL) data for 4,907 proteins. Intersection of the retrieved data with the previously reported 6,888 druggable genes yielded 2,962 druggable proteins encoded by druggable genes. Subsequently, cis-pQTLs were selected as the exposure variable, including genes from the 1 Mb upstream and downstream regions.
All the genetic exposure data were derived from the European populations, and are presented in Table 1.
Table 1
Characteristics of included GWAS data
Instrumental variable selection
Genetic variation data generated from GWAS were selected as instrumental variables (IVs). The selected IVs met the following three criteria [16]: 1) they are significantly associated with the exposure factors; 2) they should not be linked to the confounders of outcomes; 3) they affect outcomes only through exposure and not through other pathways. Therefore, we performed correlation analyses on the exposure data, setting the threshold of significance at p < 5 × 10-8. Independent exposure SNPs were selected after removal of linkage disequilibrium (LD) based on r2 < 0.1 and kb =10,000 [17, 18]. The F-test excluded weak IVs with an F-value > 10.
Outcome data
Datasets containing the genetic variations in LF, cirrhosis, and HCC were used as outcome variables and were obtained from the FinnGen Consortium (https://www.finngen.fi/en) – a large-scale Finnish genomic project established in 2017 to integrate genomic data with digital health records from 500,000 Finns. A key strength is leveraging Finland’s unique genetic history: its relatively isolated population harbors distinct genetic variations compared to other Europeans. This study utilized outcome data from FinnGen release R11; for LF, there were 188 cases and 448,864 controls; for cirrhosis, there were 1,470 cases and 448,864 controls; and for HCC, there were 609 cases and 345,118 controls. Cases in the LF, cirrhosis, and HCC cohorts were identified using ICD codes from the Finnish National Health Registry (covering hospitalizations, outpatient visits, medications, and mortality records). The general cohort comprised individuals without the specific target disease (LF, cirrhosis, or HCC) but potentially having other conditions, reflecting a more realistic population genetic background. Participants had a relatively high median age (63 years), and a large proportion were recruited from hospitals. Table 1 provides detailed descriptions of the study data.
Statistical analysis
Two-sample Mendelian randomization
In the MR analysis, the Wald ratio method was calculated for IVs with one SNP [17], and for IV with two or more SNPs, the inverse variance weighted (IVW) was employed to determine the causal relationships between the exposure and outcomes [19]. To adjust for multiple testing, we adopted the Bonferroni correction, setting a threshold p value at 1.13E -05 (0.05/4,439) for gene-level analysis and 1.68E -05 (0.05/2,962) for protein-level analysis. The MR-Egger intercept test was performed to explore the existence of pleiotropy [20], whereas the Cochran’s Q test was conducted to test for heterogeneity. The p values in the above two analyses were set at > 0.05. To test the stability of the results, the leave-one-out method was employed to carry out sensitivity analyses.
Colocalization analysis
For candidate drug targets found to be significantly causally associated with liver diseases (LF, cirrhosis, and HCC), we further performed colocalization analyses to identify common genetic variants between them and liver diseases. The following posterior probabilities for the five hypotheses were derived from the colocalization analysis [21]: PPH0, which was not associated with either candidate drug targets or diseases; PPH1, which was associated with the candidate drug targets but not with diseases; PPH2, which was associated with diseases but not with the candidate drug targets; PPH3, which was associated with both candidate drug targets and diseases with different causal variants; PPH4, which was associated with both candidate drug targets and diseases, with the common causal variant. PPH4: 0.5-0.8 reflected moderate evidence of colocalization, and PPH4 > 0.8 reflected high evidence [22]. The candidate drug targets that were colocalized with the diseases were considered significant candidate drug targets.
All analyses were performed using the packages “TwoSampleMR”, “Mendelian Randomization”, and “coloc” in the R software (version 4.3.2).
Supplementary validation
Additional validation tests for causality of significant candidate targets with LF, cirrhosis and HCC were conducted using summary-data-based MR (SMR) [23], based on data from eQTLGen and GTEx (https://www.gtexportal.org/). The heterogeneity in dependent instruments (HEIDI) test was employed to differentiate causal associations between genes and diseases from common genetic variants or linkage disequilibrium [13]. For SMR, p < 0.05 was set as the threshold of statistical significance; for the HEIDI test, p < 0.05 was set as the threshold of statistical significance for the causal association that was driven by linkage disequilibrium. These analyses were performed using the SMR v1.3.1 package. The details of SMR data are presented in Table 1.
Preliminary exploration of regulatory mechanisms
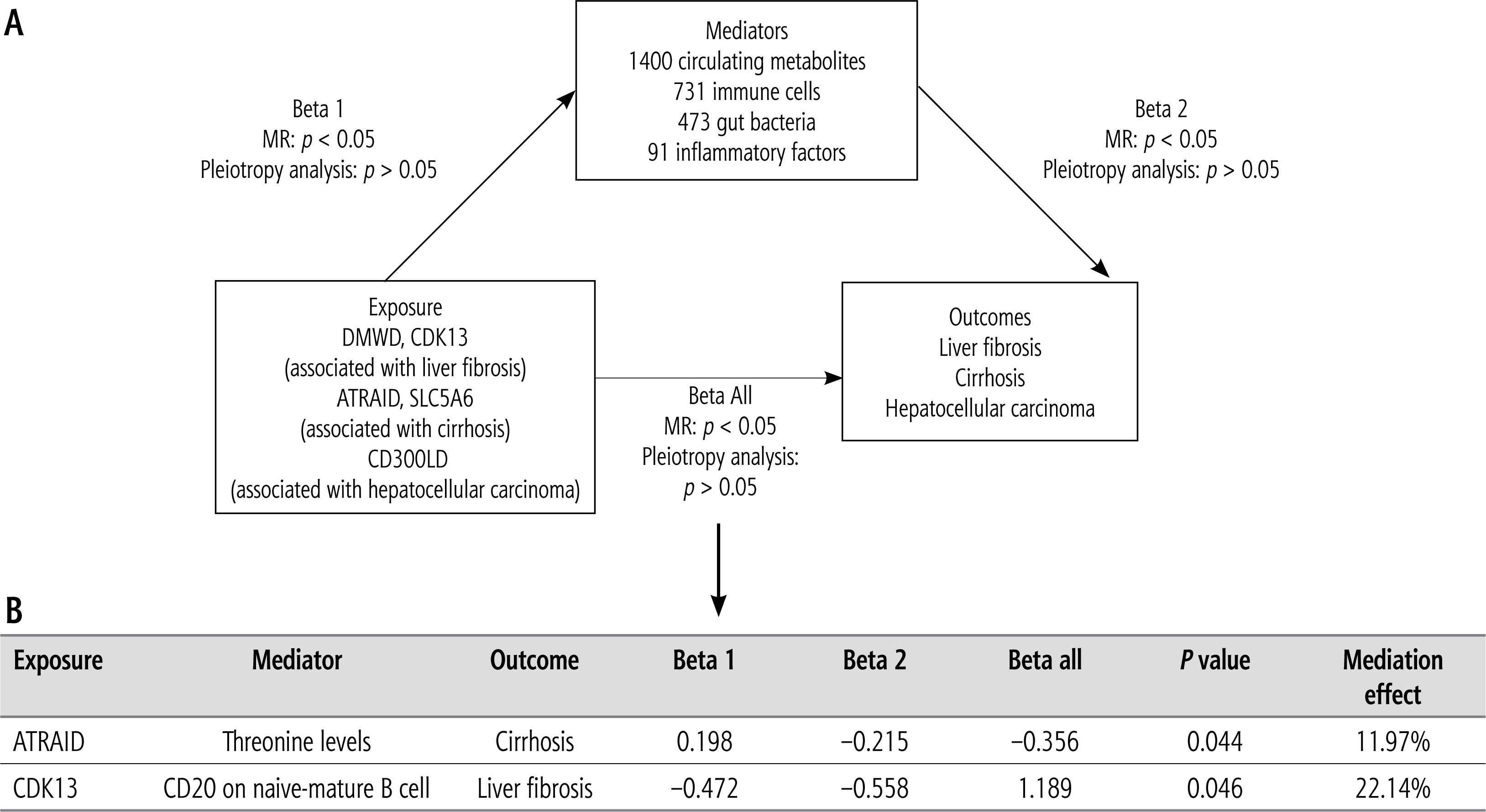
To predict the pathways involved in the mechanisms of disease modulation by the identified candidate targets, we performed a two-step MR. The mediating variables were 1400 circulating metabolites (GCST-90199621–GCST90201020) [24], 731 immune cells (ebi-a-GCST90001391–ebi-a-GCST90002121) [25], 473 gut microbiota (GCST90032172–GCST0032644) [26], and 91 inflammatory proteins (GCST90274758–GCST90274848) [27]. Their sources are detailed in Table 1. The above data were analyzed and screened using similar criteria as for the two-step MR. The cau-sal effect between exposure and outcome variables was calculated as the total effect (Beta all), followed by determination of the causal effects between the exposure and mediating variables (Beta 1), and between the mediating and outcome variables (Beta 2). Finally, the indirect effects, also known as the mediator effects (Beta 1 * Beta 2) and direct effects (Beta all – Beta 1 * Beta 2) were calculated [28].
Results
Two-sample MR identified candidate therapeutic targets
The two-sample MR analysis revealed 9 druggable genes, which were significantly causally associated with LF and considered as candidate therapeutic targets for LF. These targets included ERCC3, PISD, RABEP1, P2RX4, BCR, SENP6, CA6, DMWD, and CDK13 (Fig. 2A, Supplementary Table 1). Among them, ERCC3, PISD, P2RX4, BCR, SENP6, CA6, and CDK13 were associated with increased risk of LF, whereas RABEP1 and DMWD decreased the risk. Supplementary Table 1 demonstrates that the p values of Cochran’s Q test and the MR-Egger intercept test were > 0.05, suggesting that none of the targets had heterogeneity and pleiotropy. Supplementary Figure 1 display the results of sensitivity analysis, indicating that, after each SNP was removed, the MR analysis was re-performed and the results were not significantly altered, confirming that the results were stable.
Fig. 2
Results of Mendelian randomization analysis of LF, cirrhosis and HCC based cis-eQTL and cis-pQTL. A) Forest plot of 9 candidate targets for LF. B) Forest plot of 6 candidate targets for cirrhosis C) Forest plot of 12 candidate targets for HCC. Multiple inspection criteria: p value < 0.05/4,439 = 1.13E-05 (cis-eQTL), p value < 0.05/2,962 = 1.68E-05 (cis-pQTL)
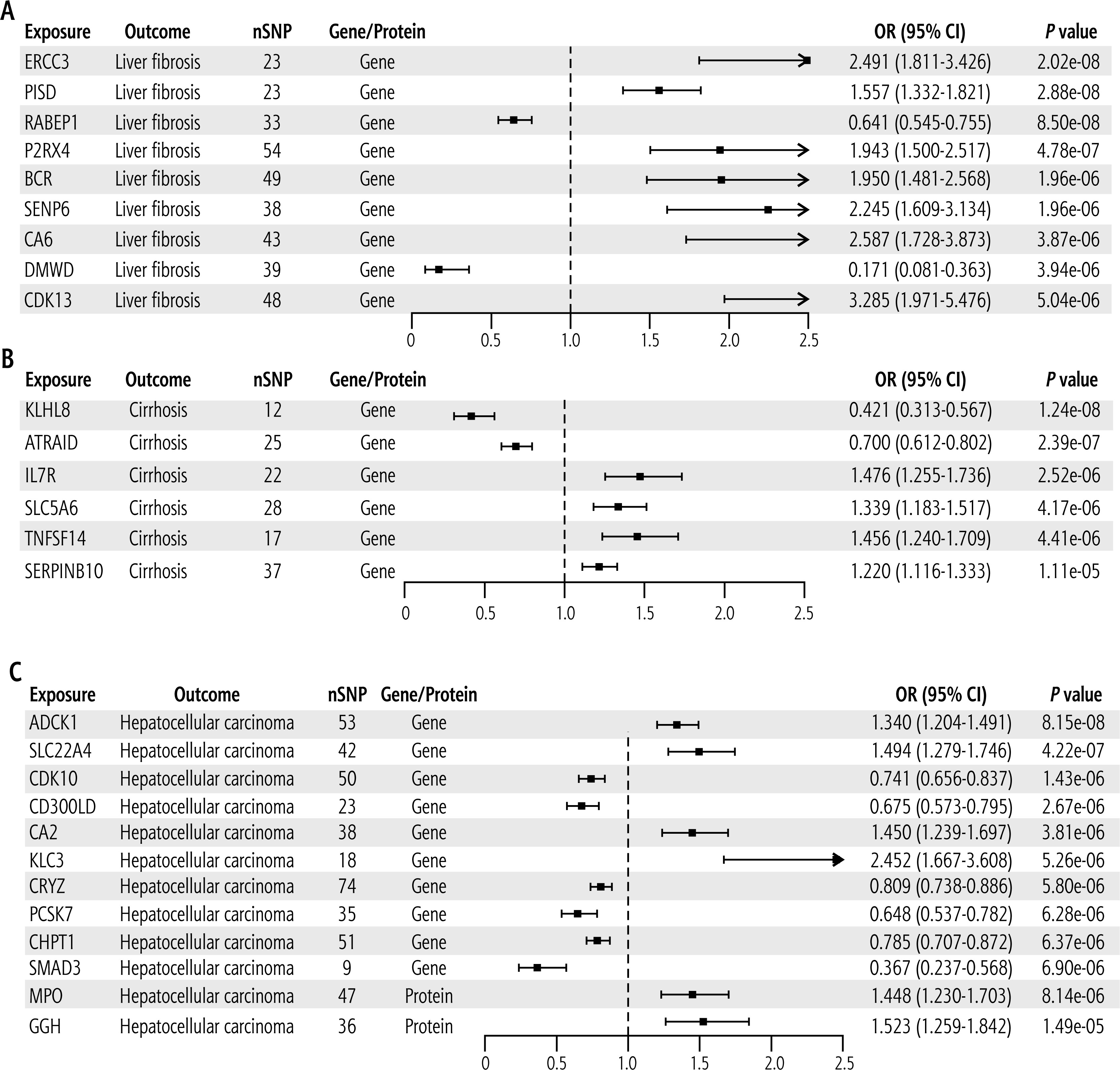
Six druggable genes that were causally associated with cirrhosis were identified, with KLHL8 and ATRAID reducing the risk of cirrhosis, while IL7R, SLC5A6, TNFSF14, and SERPINB10 were predicted to increase the risk (Fig. 2B, Supplementary Table 2). These targets had no heterogeneity and pleiotropy (Supplementary Table 2), and sensitivity analyses confirmed that the results were stable (Supplementary Fig. 2).
For HCC, 12 candidate therapeutic targets were identified, including 10 druggable genes and 2 druggable proteins. Among the genes, ADCK1, SLC22A4, CA2, and KLC3 were associated with increased risk of HCC, whereas CDK10, CD300LD, CRYZ, PCSK7, CHPT1, and SMAD3 were associated with reduced risk. The two druggable proteins MPO and GGH increased the risk of HCC (Fig. 2C, Supplementary Table 3). Notably, all these candidate therapeutic targets had no significant heterogeneity or pleiotropy (Supplementary Table 3), and sensitivity analyses confirmed that the results were robust (Supplementary Fig. 3).
Table 2 displays the above candidate targets and their official full names.
Colocalization analysis identified significant candidate therapeutic targets
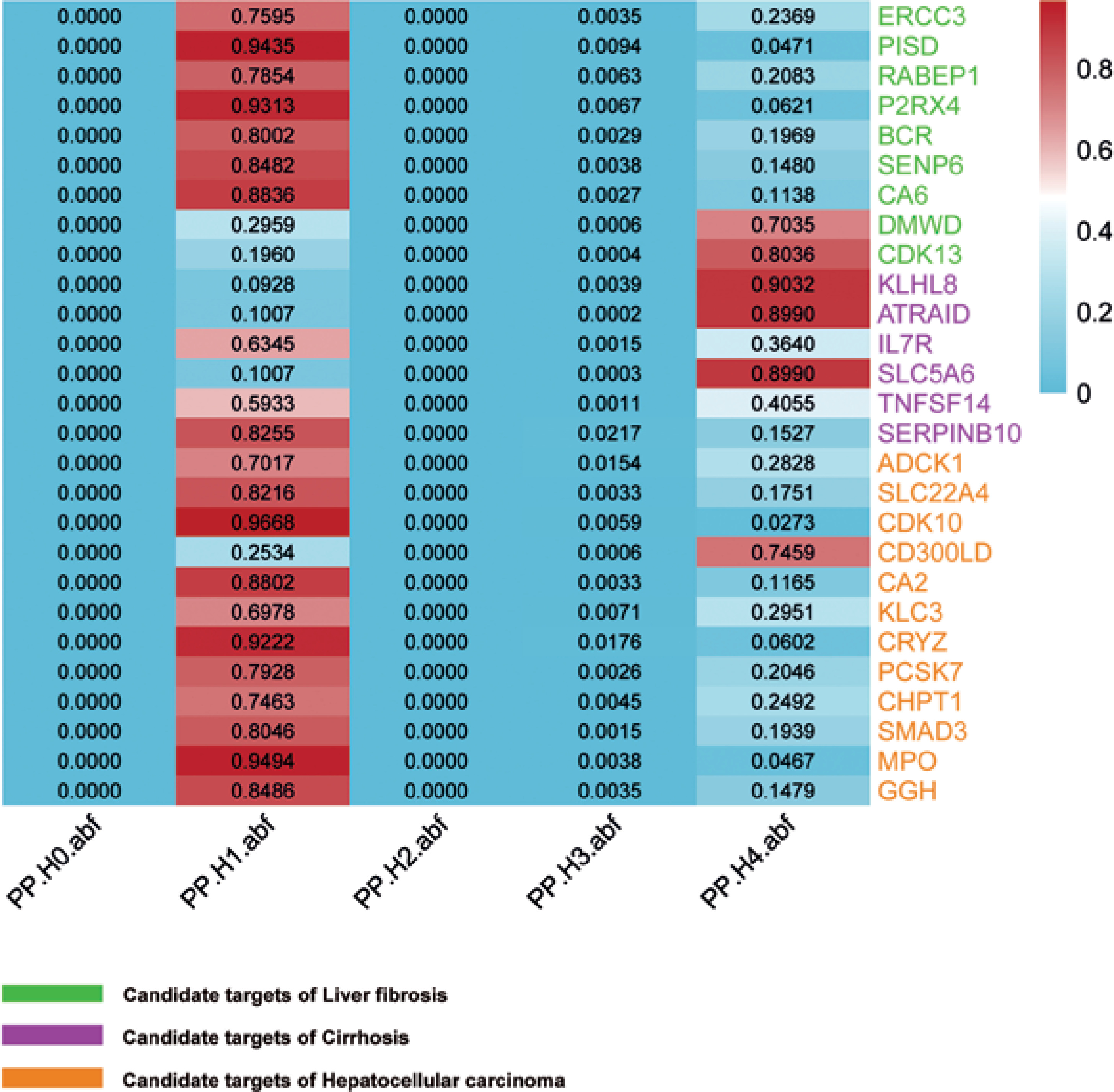
The colocalization analysis identified 2 candidate therapeutic targets (DMWD and CDK13) sharing genetic variants with LF, 3 candidate therapeutic targets (KLHL8, ATRAID, and SLC5A6) sharing genetic variants with cirrhosis, and 1 candidate therapeutic target (CD300LD) sharing genetic variants with HCC. Colocalization analysis further revealed causal relationships between the targets and diseases, and 6 targets were considered to be significant candidate targets for liver diseases. The detailed colocalization analysis results are shown in Figure 3.
Fig. 3
Colocalization analysis for the significant candidates. DMWD and CDK13 share genetic variants with LF and are significant candidate targets for LF; KLHL8, ATRAID, and SLC5A6 have common genetic variants with cirrhosis and are significant candidate targets for cirrhosis; CD300LD shows common genetic variation with HCC and is a significant candidate target for HCC (Median colocalization support: PPH4: 0.5-0.8. High colocalization support: PPH4 > 0.8)
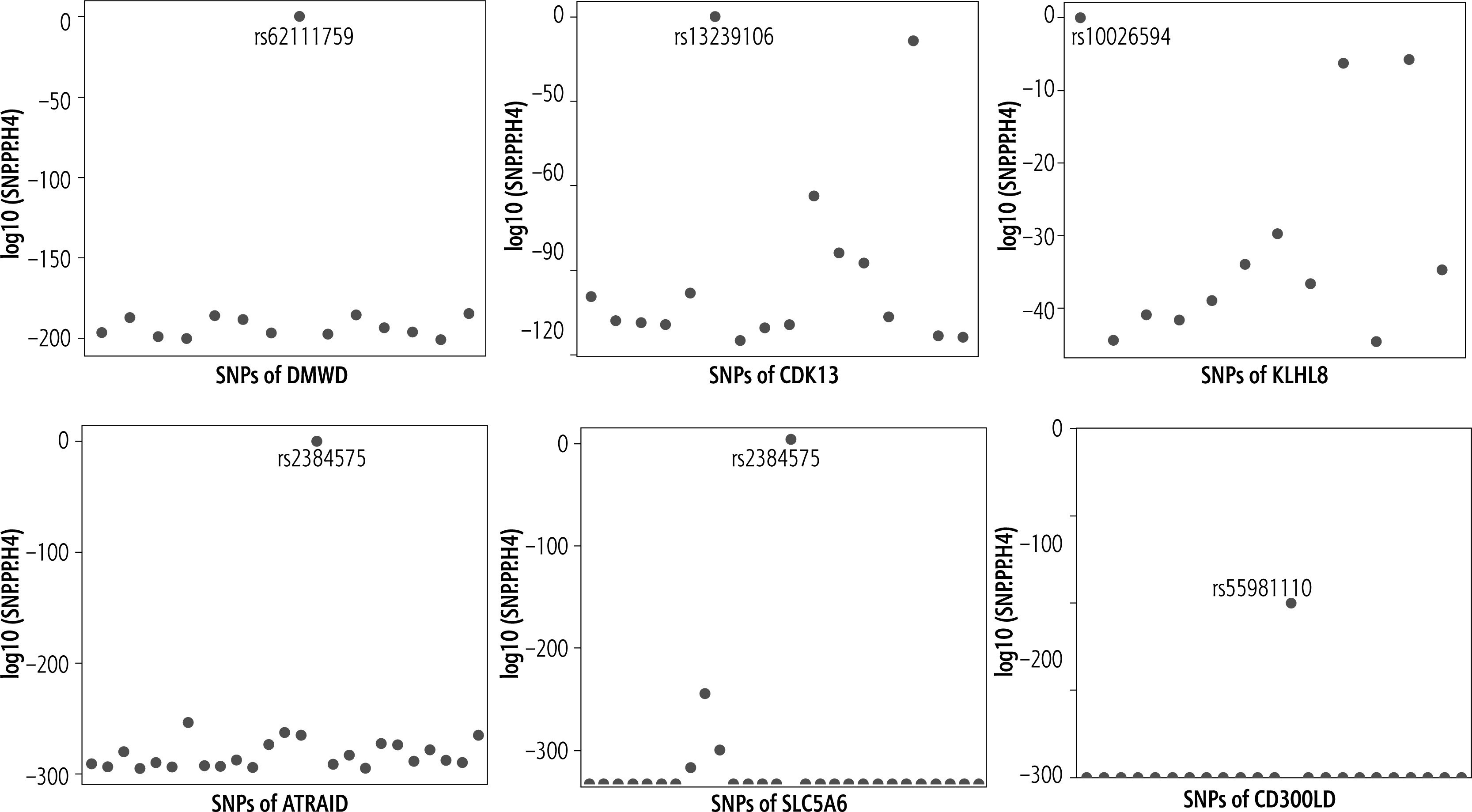
Further analysis revealed that DMWD shared the genetic variant SNP rs6211759 with LF (Fig. 4A), CDK13 and LF shared the common genetic variant rs13239106 (Fig. 4B); KLHL8, ATRAID, and SLC5A6 shared genetic variants rs10026594, rs2384575 and rs2384575 with cirrhosis (Fig. 4C-E); rs55981110 was the common genetic variant of CD300LD and HCC (Fig. 4F).
Fig. 4
Common genetic variants for significant candidate targets and diseases. A) Rs62111759 is the common SNP of DMWD and LF; B) Rs13239106 is the common SNP of CDK13 and LF; C) Rs10026594 is the common SNP of KLHL8 and cirrhosis; D) Rs2384575 is the common SNP of ATRAID and cirrhosis; E) Rs2384575 is the common SNP of SLC5A6 and cirrhosis; F) Rs55981110 is the common SNP of CD300LD and HCC
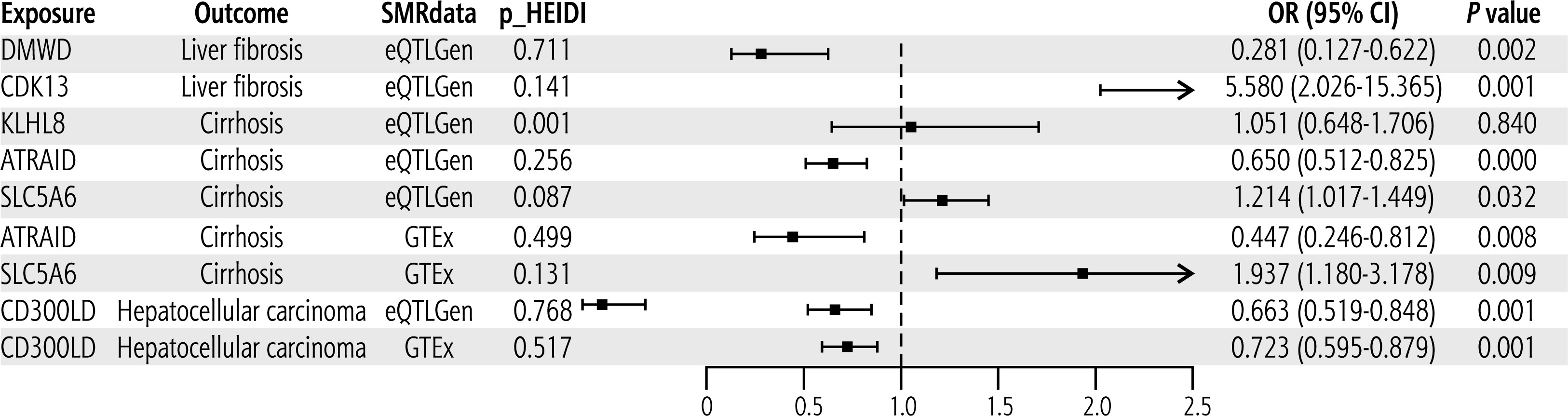
Supplementary validation for significant candidate therapeutic targets
Through the SMR analysis, we validated the causal association of significant candidate targets with liver diseases using two data sets (eQTLGen and GTEx). The data volume in GTEx is relatively small, and hence we could only validate the results of ATRAID, SLC5A6, and CD300LD in the GTEx group. The validation results are shown in Figure 5. In the eQTLGen group, DMWD was predicted to reduce the risk of LF, whereas CDK13 increased the risk of LF, ATRAID reduced the risk of cirrhosis, while SLC5A6 increased the risk of cirrhosis, CD300LD reduced the risk of HCC, and there was no causal relationship between KLHL8 and cirrhosis. Except for KLHL8, the associations of the other targets with liver diseases were consistent with the results of the two-sample MR analysis. In the GTEx group, ATRAID was predicted to reduce the risk of cirrhosis, while SLC5A6 increased the risk of cirrhosis, and CD300LD reduced the risk of HCC. These results were consistent with the two-sample MR and the results obtained for the eQTLGen group. Data shown in Figure 5 indicate that the p values of the HEIDI test for all targets in the two data sets, except KLHL8, were greater than 0.05, indicating that the causal relationship between these targets and liver diseases was driven by the common genetic variation rather than linkage disequilibrium.
Preliminary exploration of regulatory mechanisms
To identify the regulatory mechanisms, a two-step MR was performed for significant candidate targets DMWD, CDK13, ATRAID, SLC5A6, and CD300LD. As shown in Figure 6A, mediation analysis was performed with 1400 circulating metabolites, 731 immune cells, 473 gut microbiota, and 91 inflammatory proteins. Among the circulating metabolites, ATRAID was predicted to inhibit cirrhosis by upregulating threonine (p = 0.044, mediation effect: 11.97%) (Fig. 6B, Supplementary Table 4). Moreover, CDK13 was found to promote liver fibrosis by inhibiting CD20 on naive-mature B cells (p = 0.046, mediation effect: 22.14%) (Fig. 6B, Supplementary Table 5). However, none of 473 gut microbiota and 91 inflammatory proteins was involved in the regulation of liver diseases by the 5 targets (Supplementary Tables 6 and 7).
Discussion
In this study, we identified 9 candidate targets for LF, 6 candidate targets for cirrhosis and 12 candidate targets for HCC through the two-sample MR. Colocalization analysis uncovered 2 significant candidate targets for LF, 3 significant candidate targets for cirrhosis, and 1 significant candidate target for HCC. The results of the supplementary validation analysis using SMR were consistent with those of the two-sample MR, except for KLHL8. Finally, two-step MR was employed to explore the regulatory mechanisms of the validated targets, which revealed that ATRAID inhibited cirrhosis by reducing the levels of threonine, and CDK13 was predicted to promote LF by inhibiting CD20 on naive-mature B cells. To our knowledge, this is the first study to identify the therapeutic targets of LF, cirrhosis, and HCC, as well as their regulatory mechanisms through dual-omics.
For the LF, we identified 9 candidate targets (ERCC3, PISD, RABEP1, P2RX4, BCR, SENP6, CA6, DMWD, and CDK13). Ding et al. reported that activation of the EGF-EGFR/CHKA/PI3K-PDK1-AKT signaling cascade enhances the expression of ERCC3-regulated oncogenes to promote the progression of nonalcoholic fatty liver disease to HCC [29]. Moreover, the ERCC3 gene was predicted to increase the risk of LF; however, further investigations are needed to clarity whether ERCC3 can enhance the progression of LF to HCC. Ma et al. found that the exosome circCDK13 can inhibit LF via the miR-17-5p/KAT2B axis, and our study showed that increased expression of CDK13 may promote LF, and thus there may be a mechanistic link between the two findings that need to be further explored [30]. None of the remaining targets have previously been reported to be associated with liver fibrosis. Furthermore, we performed colocalization analysis to identify the significant candidate targets DMWD and CDK13, both of which were confirmed by the SMR method. In the analysis of regulatory mechanisms of the two targets, we found that CDK13 may promote liver fibrosis by inhibiting CD20 on naive-mature B cells. Numerous studies have shown that B cells are associated with the occurrence and development of LF [31–33], and the present findings reveal a new mechanism for the role of the immune system in LF development.
For cirrhosis, 6 candidate targets (KLHL8, ATRAID, IL7R, SLC5A6, TNFSF14, and SERPINB10) were identified by the two-sample MR. Ex vivo experiments demonstrated that the IL7R expression accelerated the progression of cirrhosis [34], which is consistent with our finding. Elsewhere, Mells et al. identified IL7R as a genetic risk factor for the occurrence of primary biliary cirrhosis [35]; therefore, IL7R is likely to be an important target for cirrhosis. A previous study showed that splenectomy could effectively reduce the expression level of TNFSF14 in the blood, thereby reducing liver fibrosis and cirrhosis in patients [36]. Notably, TNFSF14 has been linked to the development of fibrosis in several organs, including the heart, lung, and kidney [37–39]. Therefore, we speculated that TNFSF14 may be a promising candidate target for investigation of cirrhosis. To our knowledge, no study has explored the association of KLHL8, ATRAID, SLC5A6, and SERPINB10 with cirrhosis or liver fibrosis. Here, three candidate targets – KLHL8, ATRAID, and SLC5A6 – were identified through colocalization analysis, among which ATRAID and SLC5A6 are considered to be significant genes. The preliminary analysis of the target mechanisms revealed that ATRAID may inhibit cirrhosis by reducing threonine levels; however, this result needs to be further validated through basic experiments.
As for HCC, 12 candidate targets (ADCK1, SLC22A4, CDK10, CD300LD, CA2, KLC3, CRYZ, PCSK7, CHPT1, SMAD3, MPO and GGH) were identified in the preliminary analysis. Zhong et al. reported that decreased CDK10 expression was positively associated with the occurrence and progression of HCC [40], which is consistent with our findings. Overexpression of SMAD3 mediates the inhibitory effect of transforming growth factor β1 (TGF-β1) on the catalytic subunits, thereby promoting lipid peroxidation in HCC cells [41], whereas NOP2/Sun RNA methyltransferase 5 has been reported to accelerate the progression of HCC by upregulating SMAD3 expression [42]. TRIP13 silencing as a tumor suppressor in HCC can inhibit cell growth and metastasis via activating the TGF-β1/SMAD3 signaling pathway [43], therefore regulating HCC through the SMAD3 complex. Available studies have demonstrated that MPO overexpression favors HCC progression [44, 45], which is consistent with our findings. Among them, ADCK1, SLC22A4, CD300LD, CA2, KLC3, CRYZ, PCSK7, CHPT1, and GGH are new unreported targets in HCC. Further analyses showed that CD300LD was a key target for driving cancer immunotherapy [46, 47]. This indicates that further research is needed to explore the therapeutic potential of CD300LD in HCC.
In this study, novel therapeutic targets for LF, cirrhosis, and HCC were revealed by MR with dualomics analysis at the gene and protein levels. No evidence of pleiotropy or heterogeneity was found in all targets analyses, and sensitivity analyses suggested that all results were stable. To predict the therapeutic value of the targets, we used genetic variation data of druggable genes and druggable proteins as exposure variables, from the latest and most comprehensive datasets. Next, colocalization analyses were conducted to identify the common genetic variants driving the targets and diseases. Additional validation of the targets was also carried out using SMR from multiple sources. This led to the identification of CDK13 and ATRAID as key targets influencing LF and cirrhosis through immune cells and metabolites, providing ideas for the development of new targeted drugs.
This study has several limitations. Given the limitations of accessing original data, all participants were European; therefore, the conclusions may not be applicable to other populations. Second, the proteomic data were smaller in scale compared to genomic data, and therefore fewer protein targets were identified. Thirdly, the targets and mechanisms identified in this study need to be further explored in basic experiments.
Conclusions
In this study, novel therapeutic targets for LF, cirrhosis, and HCC were identified through dual-omics analysis, colocalization, and validation. The downstream regulatory mechanisms were probed, with CDK13 and ATRAID identified for LF and cirrhosis. This study provides important reference data to guide future investigations into disease mechanisms and the development of new targeted drugs.





