
Introduction
Inflammasomes constitute a part of the innate immune system, responsible for protection against pathogens [1]. They are intercellular, multimeric protein complexes consisting of nucleotide-binding oligomerization domain receptors (NLR), apoptosis-associated speck-like protein containing a CARD (ASC), and an enzymatic effector – caspase. Its role involves catalysing protective reactions of the body by the generation of cytokines and death of infected cells in the apoptotic mechanism [2]. In the last few years, studies have been increasingly focused on inflammasomes and their role in the pathogenesis of diseases of the cardiovascular [3], respiratory, or gastrointestinal systems [4].
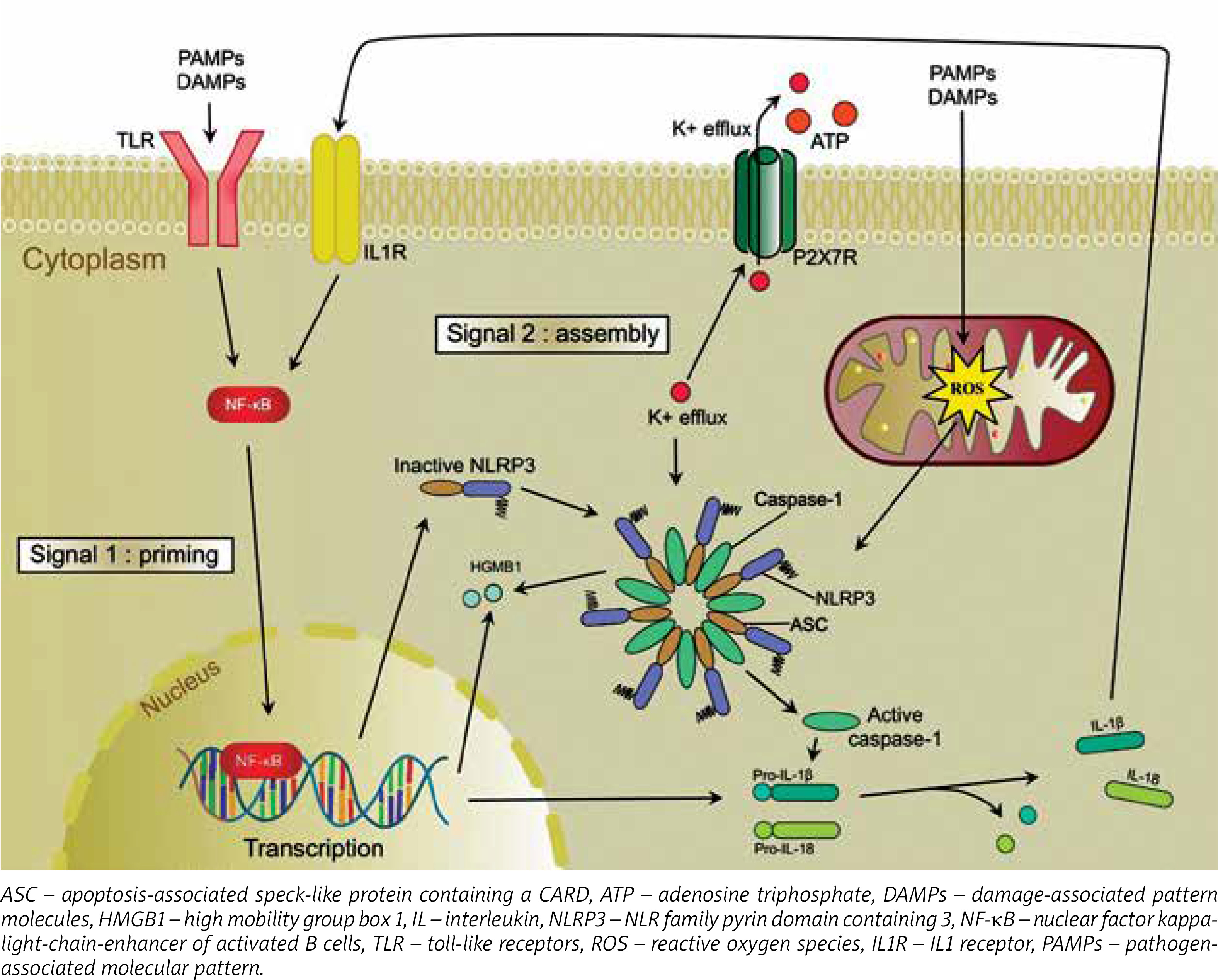
Inflammasomes become activated when a pattern recognition receptor (PRR) detects conservative structures of microorganisms, known as pathogen-associated molecular pattern (PAMPs) and damage-associated pattern molecules (DAMPs). It leads to activation of signalling pathways, which trigger antibacterial inflammatory response.
Pattern recognition receptors can be classified into 3 main classes: toll-likely receptors (TLR), retinoic acid-inducible gene I (RIG-I) receptors, and NOD-like (NLR) receptors [5].
Of all receptors belonging to the NLRP sub-family, the NLRP3 receptor is the best described in terms of its role in pathophysiology and clinical implications [6, 7]. The activation of the NLRP3 inflammasome requires 2 signals (Figure 1). It results in the activation of caspase-1, which controls maturation and release of the proinflammatory cytokines interleukin (IL)-1β and IL-18. Production of cytokines is a direct response to cell damage or recognition of infection factors [1].
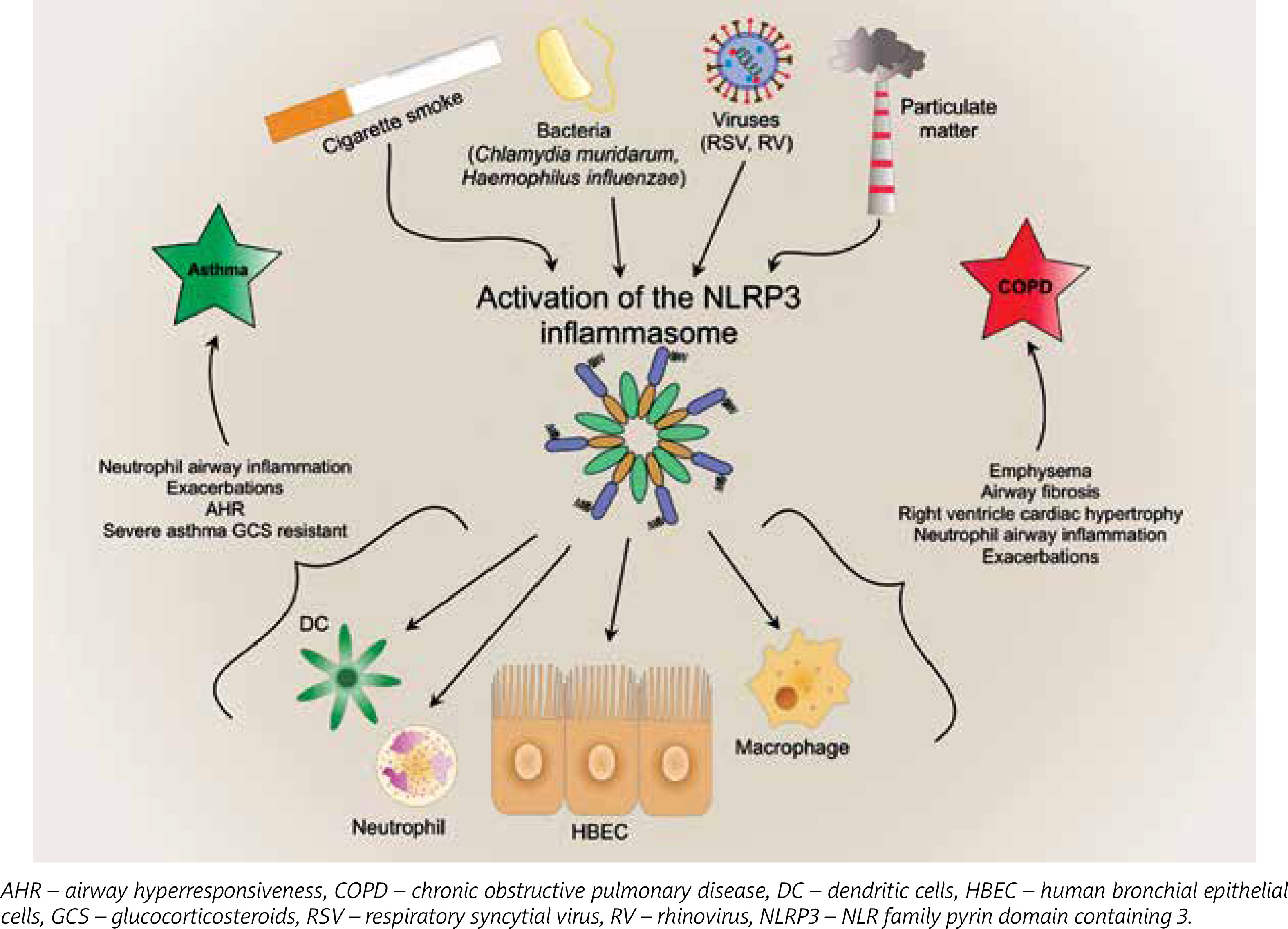
Interleukin 1β, being the main proinflammatory cytokine, plays a role in triggering and maintaining inflammation of airways by inducing the release of many cytokines, e.g. IL-2, -3, -4, -5, -6, -8, INF-γ, and TNF. Additionally, IL-1β induces leukocytosis by release of neutrophils from bone marrow [8] and is considered a key driver of neutrophil airway inflammation in chronic obstructive pulmonary disease (COPD) [9] and asthma [10] (Figure 2). Furthermore, it activates eosinophils in asthma. Eosinophils, by releasing major basic protein (MBP), contribute to overstimulation of M3 receptors by acetylcholine, which results in airway smooth cell (ASM) contraction and overproduction of mucus [10]. Interleukin 18 plays an important role in type 1 T helper cells (Th1)/type 2 T helper cells (Th2) polarization. Some studies on animals models confirm the role of the interleukin in induction of emphysematous lesions, airway fibrosis, mucus metaplasia, as well as right ventricle cardiac hypertrophy, which might be one of the most severe complications of COPD [11, 12]. Interestingly, IL-18 knock-out mice were protected against cigarette smoke (CS)-induced inflammation and emphysema, which indicated a pivotal role of this cytokine in the pathogenesis of COPD [13]. Overexpression of IL-18 protein in the lungs induces type 1 and type 2 cytokines and airway inflammation, and results in increasing airway hyperresponsiveness via CD4+ T cells and IL-13 in asthma [14].
Figure 2
The effects of activation of inflammasome in asthma and chronic obstructive pulmonary disease
NLRP3 can be activated by many factors, such as microbiological exogenous stimuli, which, among others, can contain lipo-saccharides, lipo-oligosaccharides, nucleic acids, MDP (muramyl dipeptide), and certain bacterial pore-forming toxins, such as pneumolysin [15–19].
Of the many factors which trigger the activation of NLRP3, 3 mechanisms that probably explain this process have been identified.
According to the first hypothesis, reactive oxygen species (ROS), which are generated in the metabolic process of microorganisms penetrating body cells, might play a crucial role in NLRP3 activation [20].
The second hypothesis implies that NLRP3 activation may be associated with intracellular decreased level of K+ ions, being a result of a malfunction of the potassium channel, or may be associated with bacterial toxins, inducing pores in the cell membrane, which results in an efflux of K+ ions from the cell [21].
In the third option, NLRP3 is activated via lysis of the cell membrane and release of cathepsin B-likely protein or protein modified by cathepsin B into cell cytosol, which is the result of a phagocytized microorganism [22].
It was also hypothesized that efflux of K+ ions from the cell, damage to the cell membrane, or the presence of ROS might oxidize mitochondrial DNA in the cell, which results in NLRP3 activation [23].
Chronic obstructive pulmonary disease
Chronic obstructive pulmonary disease is characterized by high prevalence and high mortality. It is estimated that by the year 2030, this disease will have become the third most common cause of death [24]. It manifests with progressive and irreversible obturation, persistent infection of airways, and gradual depletion of the lung function. The intensity and occurrence of particular symptoms (such as cough, dyspnoea) depend on the degree of one of two classic phenotypes: emphysema or chronic bronchitis, which results in different manifestations of the disease in different patients [25]. Infections, mostly of bacterial aetiology, contribute to periodical exacerbations, which often require hospitalization and modified treatment. Chronic inflammation of the airways is caused by inhalation of noxious particles, most often CS – about 20% of tobacco smokers develop this condition [26]. It was revealed that CS activates NLRP3 inflammasome, which occurs mainly due to ROS found in it, and which are also released from mitochondria by CS [27, 28]. ROS generation is modulated by efflux of K+, another crucial factor required in NLRP3 activation [29]. Moreover, extracellular ATP (eATP) is released, then by binding to the P2X7 receptor it activates the caspase-1/NLRP3 pathway. This observation was confirmed on mouse models [30]. It was confirmed that ATP accumulates in the airways of animals and patients with COPD, which implies a crucial role of this molecule in the pathogenesis of COPD [31]; likewise, the P2X7 receptor is upregulated on alveolar macrophages and blood neutrophils from patients with COPD [32]. It is widely known that CS contributes to a release of endogenous danger signals. One of them is high mobility group box 1 (HMGB1) protein, which belongs to the family of DAMPs. By interacting with TLR4, it can activate inflammasome. Studies on this protein found in BAL fluid and sputum of patients with COPD point to its role in the above disease [29, 33].
Finally, all those factors lead to assembly of inflammasome complex and activation of caspase-1, which cleaves pro-IL-1β and pro-IL-18 and contributes to a release of their active forms from, among others, human bronchial epithelial cells (HBEC), and macrophages. Mortaz et al. showed that NLRP3 activation from HBEC might result in a release of chemokine ligand 8 (CXCL-8/IL-8) [34], also known as neutrophil chemotactic factor, responsible for migration of neutrophils to airways, secreting elastases and proteins (such as IL-8, matrix metallo-proteinase [MMP]-9, neutrophil elastase, proteinase-3), which are the main mediators of the tissue damage and unceasing depletion of the lung function in this condition [35, 36]. Nevertheless, literature regarding the involvement of the caspase-1/NLRP3 axis in releasing CXCL8 is still discordant. A study conducted on an in vitro model a few years later revealed that NLRP3 silencing does not affect the level of this protein [37].
There are abundant scientific reports implying that activation of NLRP3 inflammasome signalling pathway participates in the development of COPD, mainly in animal models [38–40]. It has been shown that mice exposed to CS presented pathophysiological characteristics of COPD, whereas NLRP3-knock out mice did not demonstrate any significant changes in lung function [41].
Furthermore, not only CS but also biomass fuel smoke (BS), being the main risk factor in low-income countries, participate in the development of chronic inflammation in COPD. This type worsens quality of life and is characterized by predominance of chronic bronchitis [25]. BS contains both PAMPs and DAMPs, including ROS. They induce intracellular oxidative stress and activate NLRP3 in a way similar to that of CS [37, 42].
Some recent studies revealed that NLRP3 inflammasome plays a crucial role in the development of acute exacerbations of COPD (AECOPD). One of the first such reports was published in 2014. Its authors noticed a potential role of IL-27, IL-37, and PYD domains containing protein 7 (NALP7) in the progression of stable COPD and, what is particularly important , a lack of correlation regarding NLRP3 inflammasome in these patients, in whom it remained inactive [43]. In support of this, one study found the presence of pre-assembled ASC specks in bronchoalveolar lavage fluid (BALF) in stable COPD patients in contrast to healthy donors, as well as their accumulation in the lungs [44], which implies inactivity of NLRP3 in patients without exacerbations. Briefly, in clinically stable patients, NLRP3 inflammasome is primed, but not activated; it becomes activated in infectious AECOPD [45]. This relationship was also demonstrated in an in vitro model of lipopolysaccharide (LPS)-induced exacerbations [46]. More frequent exacerbations are associated with an increase in systemic inflammation and airway level of IL-1β, which, by binding with IL1 receptors (IL1-R), activates NLRP3, perpetuating an immune response [29, 47]. It results in a faster decline in the lung function and poorer quality of life. It has been demonstrated that NLRP3 is involved in exacerbations of bacterial aetiology, and this type of exacerbation is the predominant one. There is positive correlation between mRNA of NLRP3-related proteins (Casp-1, ASC, IL-18, IL-1β) and the presence of 6 common pathogens of the lower respiratory tract [48]. Several studies indicate that the level of IL-1β in sputum can serve as a marker of bacterial exacerbations [9, 49].
It has been also observed that caspase-1 not only induces the release of IL-1β and IL-18 but also IL-1α from peripheral blood mononuclear cells (PBMCs) in unstable/exacerbated COPD patients in the absence of melanoma 2 (AIM2)/caspase-1/caspase-4 dependent pathway [50]. In support of this, Pauwels et al. showed that lung tissue and induced sputum of patients with COPD contain increased levels of IL-1β and IL-1α [38]. Both IL-1α and IL-1β play an important role in fibrosis [51]. Additionally, activation of AIM2 inflammasome and release of IL-1α results in secretion of transforming growth factor β (TGF-β), which is also responsible for lung fibrosis in COPD [50].
Asthma
Asthma is one of the most common respiratory disorders in the world. It is suggested that approximately 300 million people suffer from asthma worldwide [52]. This number is estimated to increase to 400 million by the year 2025 [53].
Typical symptoms of asthma include the following: recurrent episodes of wheezing, breathlessness, tightness in the chest, and cough. They are accompanied by bronchial hyperreactivity and airway obstruction. However, in some patients, the reversibility of airflow limitation may be incomplete [54]. Recent studies focused on inflammatory aspects of asthma development, demonstrating a crucial role of inflammasome-dependent processes. The vast majority of studies regarding inflammasomes in asthma are based on animal models of the disease, and only a few studies have undertaken this issue in patients. Jodie et al. noted that gene expression for NRLP3 inflammasome components was significantly increased in the sputum of patients affected by neutrophilic asthma in comparison with healthy subjects and patients affected by eosinophilic and paucigranulocytic asthma. Furthermore, gene expression of proinflammatory caspases 1, 4, and 5 was also elevated. The researchers also noted the presence of the mature form of IL-1β in supernatants of patients with neutrophilic asthma after application of the western blot technique. Expression of receptors, such as NOD2 and TLR2, which also promote IL-1β synthesis, was elevated in patients with neutrophilic asthma. Proteins for NLRP3 and caspase-1 were expressed in sputum macrophages in all asthma subtypes, but there was also expression in neutrophils from patients with neutrophilic asthma [55].
Results of other recently published studies imply that hyperactive NLRP3 inflammasome as well as synthesis and secretion of IL-1β are typical for neutrophilic asthma [56–58]. Lachowicz-Scroggins et al. suggest in their study that in SA, neutrophil extracellular traps (NETs) are responsible for NLRP3 activation in resident cells, such as monocytes or macrophages. Moreover, expression of IL-1R family members is elevated in the sputum of patients with severe asthma (SA) [58]. The authors also observed increased expression of NLRP3 inflammasome and an increased level of the proteins that activate it, such as C5a in patients with SA. The neutrophilic asthma type is usually more severe [59–61] and patients demonstrate higher resistance to glucocorticosteroids (GCS) [57, 60] and higher levels of NLRP3 and IL-1β mRNA in sputum [57].
Rossios et al. concluded that interleukin 1 receptor-like 1 (IL1RL1) gene expression is associated with eosinophilic SA, while NLRP3 inflammasome expression is highest in patients with neutrophilic SA [56].
The following factors: bacterial and viral infections, air pollution, allergens, and CS, may induce endoplasmic reticulum (ER) stress, which will lead to a release of DAMPs from mitochondria. This eventually results in NLRP3 inflammasome activation and conversion of pro-IL-1β into its active form [62]. Studies on mouse models aimed at investigating bacterial factors showed that Chlamydia and Haemophilus infections increase NLRP3, caspase-1, and IL-1β responses, which drive steroid-resistant neutrophilic inflammation and airway hyperresponsiveness (AHR) [57]. Increased bacterial burden in airways is positively correlated with neutrophilic inflammation in asthma, its more severe course, and resistance to GCS [63–65]. Rhinovirus (RV) infections are the main cause of asthma exacerbations in adults and children [66, 67]. NLRP3 inflammasome activation may also underlie molecular mechanisms of this phenomenon. A study on mouse models showed that RV induces TLR2-dependent inflammasome activation [68]. Earlier studies confirm NLRP3 activation and IL-1β secretion in cultured bronchial epithelial cells infected with RV [69, 70].
In U937-line cells infected with respiratory syncytial virus (RSV), activation of TLR2/MyD88 pathway (first signal) and production of intracellular ROS as well as potassium efflux (second signal) were observed. It resulted in the formation of NLRP3/ASC complex and caspase 1 activation, which subsequently cleaved pro-IL-1β protein into its mature form [71]. Not only do infection factors, such as bacteria or viruses, increase NLRP3 activation, but also functional polymorphisms in NLRP3 have been identified, which increase the stability of NLRP3 mRNA. Single nucleotide polymorphism (SNP) is associated with susceptibility to food-induced anaphylaxis and aspirin-induced asthma (AIA) [72].
Also, air pollution induces NRLP3-dependent inflammatory response. Particulate matter less than 10 μm (PM10) activates NRLP3 inflammasome in human epithelial cells, which increases IL-1β synthesis. These changes were also confirmed in the same in vivo study conducted on mouse models [42]. In PM10-induced inflammatory responses, NRLP3 knockout mice demonstrated lower concentrations of IL-1β and a lower number of inflammatory cells, particularly neutrophils, measured in BAL fluid in comparison to the wild type murine. In PM10-induced NRPL3 knockout mice only the number of macrophages was significantly higher. However, it did not correlate with increased IL-1β level.
Likewise, biopsies conducted in regional intrathoracic lymph nodes in mice, after their 24-hour exposure to PM, revealed changes in the dendritic cell (DC) phenotype. It has been suggested that those changes result from mechanisms that are dependent on the molecular complex of NRLP3 inflammasome [42].
The same group has shown that the NLRP3 inflammasome/IL-1RI axis mediates innate responses to air pollution leading to production of chemokine (C-C motif) ligand 20 (CCL-20) and granulocyte-macrophage colony-stimulating factor (GM-CSF), which are associated with dendritic cell activation and lung neutrophilia [73]. Li et al. demonstrated that the ATP/P2X7-NLRP3 axis of dendritic cells stimulates Th2 and Th17 differentiation through IL-1β and IL-18 secretion. Furthermore, activation of NRLP3 inflammasome enhances expression and release of HMGB1, which is a key molecule of innate immune response [74, 75]. These mechanisms lead to increased airway inflammation and AHR [63]. A study on a mouse model revealed that animals with deficits of NLRP3 inflammasome or caspase1/11 did not control the influx of eosinophils in airways and demonstrated increased levels of cytokines (particularly IL-33) and Th2 chemokines in response to house dust mite (HDM)-induced allergic pneumonia, which suggest that NRPL3/caspase-1 complex controls HDM-induced allergic pneumonia [76].
Scientific data regarding involvement of other inflammasomes in asthma inflammation are scarce. Recent research indicates that NLRP1 and NLRC4 inflammasomes are activated in addition to NLRP3 in the sputum of patients with neutrophilic asthma [56], and that children with asthma with gain-of-function SNPs in NLRP1 are exposed to more severe disease presentation [77].
NLRP3-inflammasome/caspase-1/IL-1β axis as a therapeutic target
In the last few years, some studies focusing on the NLRP3/caspase-1/IL-1β axis, which might become a potential therapeutic target in treatment of asthma, have been published.
A study on mouse models established that inhibition of NLRP3 signalling pathway resulted in reduced levels of IL-1β and Th2 cytokines, and what is more important, it completely inhibited HDM-induced AHR and attenuated steroid-resistant asthma [57]. Moreover, IL-1β inhibition also suppressed airway inflammation and AHR in severe, steroid-resistant (SSR) asthma. IL1-R may be a promising goal in therapy of SSR asthma. IL1-R antagonists such as Anakinra may be beneficial in therapy [57]. Studies on mouse models showed that blocking IL1-R may also have therapeutic potential in immunomodulating treatment of allergic asthma [78]. However, such a strong blockage of IL-1 or its receptors bears a risk of frequent infections of the respiratory system [57, 79]. NLRP3 inhibitors could be an effective solution because they do not affect IL-1β synthesis from other sources [57].
It has been proven that inhibition of NLRP3 inflammasome weakens most typical features of asthma and blocks Th2 and Th17 polarization as well as reducing HMGB1 expression and release from dendritic cells [63]. In a mouse model of OVA-induced allergic inflammation, the blockage of NLRP3 inflammasome was associated with a reduced number of eosinophils, decreased IL-33 synthesis, generally impaired Th2 response, decreased DC migration to regional lymph nodes, and decreased recruitment of T CD4 + cells, producing IL-13 [80]. In recent years the influence of A20 protein has been considered in the context of inflammasome functioning, particularly NLRP3, as well as in asthma. However, A20 protein has not been investigated together with inflammasome in asthma [81]. The protein A20 is encoded by an immediate early response gene and acts as a potent inhibitor of NF-κB signalling pathway [82]. Studies in animal models have demonstrated the protective effect of A20 in diseases such as arthritis, pristine-induced lupus nephritis, and type II diabetes. The activity of NRLP3 inflammasome is negatively correlated with the activity of A20 protein [83–85]. Ruiz-Gomez et al. showed that airway epithelial cells from adults with asthma express significantly less A20 [86]. Protein A20 was shown to have beneficial effects in the course of allergic asthma in a mouse model of the disease. A20 suppressed mucus production and prevented the development of AHR [87]. Particularly interesting for the involvement of A20 in asthma is its enhanced transcription of this NF-κB inhibitor following glucocorticosteroid binding to their response element in the gene, which has been observed in bronchial epithelial airway cells [88].
In COPD, similarly to asthma, the benefits derived from direct inhibition of NLRP3 are pointed out. A study on a COPD mouse model revealed that lipoxin receptor agonist BML-111 may prevent NLRP3 inflammasome activation and inhibit ROS production [89]. A similar effect may be obtained by melatonin, which attenuates inflammation through SIRT1-dependent NLRP3 inhibition [90]. Interestingly, it has been shown that 17-oxo-docosahexaenoic acid (DHA) can block NLRP3 inflammasome and inflammasome-dependent degradation of glucocorticoid receptor (GR). Therefore, the combination of GCS and 17-oxo-DHA may be beneficial [91]. Another possibility is inhibition of AIM2 inflammasome-nintedanib abrogated AIM2/IL-1α-dependent TGF-β release from PBMCs of COPD patients [48].
Conclusions
In recent years, the contribution of inflammasome-dependent signalling pathways to the development of inflammatory diseases has become the topic of multiple research studies. Numerous studies indicate that inflammasome-dependent pathways have a significant effect on induction and maintenance of inflammation in the most prevalent lung diseases, such as asthma and COPD. Better understanding of these relationships may in the future allow the development of modern therapies in the treatment of GCS-resistant SA or COPD exacerbations. Moreover, a combination of inflammasomes and A20 protein might provide much needed insight into complex molecular integrations present in asthma. Overall, inflammasome characterization and recognition of the role of A20 protein will allow identification of new therapy targets and more personal treatment options.







