
Introduction
Carcinosarcomas (CSs) are rare tumours that consist of two malignant components: carcinomatous and sarcomatous, and are often associated with poor prognosis. These neoplasms have been observed in various locations, such as uterus, ovaries, breast, gastrointestinal tract, genitourinary tract, lung, breast, bones, thyroid gland, and paranasal sinuses [1–4]. The most common site of CS is the uterus, where it is referred to as mixed malignant Müllerian tumour (MMMT). The ambivalent nature of CSs raises questions about its origin. In the vast majority of tumours, a monoclonal origin of both components was proven [5]. Comparative genetic analyses performed on various CSs (e.g. CS of parotid gland, urinary bladder, and pharynx) revealed a large overlap of chromosomal aberrations in both tumour components [6, 7].
Most authors presume that the sarcomatous component is derived from carcinoma through a process called epithelial-mesenchymal transition (EMT) [5]. Physiologically, EMT is involved in the formation of the body plan during foetal development, differentiation of tissues, and their repair. Epithelial cells undergoing EMT lose the epithelial phenotype and acquire an increased migration potential. In various cancers, partial or complete EMT occurs, thus enhancing the malignant potential [8]. The hypothesis of the crucial role of EMT in CS pathogenesis is supported by ultrastructural, immunohistochemical, and microRNA studies [9–12].
TP53 mutations seem to be crucial for CS pathogenesis. In the vast majority of CSs, concordant overexpression of p53 is found in both components [13–15]. In recent years, molecular research on uterine CS has highlighted the role of p53 and mismatch repair protein (MMR) dysfunction in its pathogenesis; however, some studies produced conflicting data, and the role of MLH1 promoter hypermethylation has not yet been evaluated in these tumours. Therefore, we investigated the p53 and MMR status in uterine CS obtained from a single institution cohort.
Material and methods
Study population
Cases of CS, recorded at the University Clinical Centre in Gdansk from 2007 to 2015, were retrieved from the archive of the Department of Pathomorphology. The microscopic slides of each case were re-evaluated by two pathologists to verify the diagnosis. Twenty formalin-fixed and paraffinembedded (FFPE) tissue blocks containing representative uterine CS samples were included in our study.
Immunohistochemistry
Tumour samples were stained with antibodies against hMLH1(Clone ES05), PMS2 (Clone EP51), hMSH2 (Clone FE 11), hMSH6 (Clone EP49), and p53 (Clone DO-7), all ready to use (DAKO, Denmark). Staining was performed on a Dakoautostainer according to the manufacturer’s instructions. The slides of all specimens were microscopically evaluated by two experienced pathologists (PC and WB). In the case of MMR proteins, nuclear staining was considered positive, and lack of nuclear staining – negative (a sign of defective MMR). The question of how to correctly interpret the immunostaining for p53 is a matter of controversy. According to the current point of view, we considered a strong/diffuse (> 75% of tumour cell nuclei) and a completely negative stain as p53 defect indicative of its mutation, (missense and nonsense, respectively), whereas a patchy/scattered pattern was regarded as a marker of normal p53 function [16]. Representative pictures are given in Figures 1 and 2.
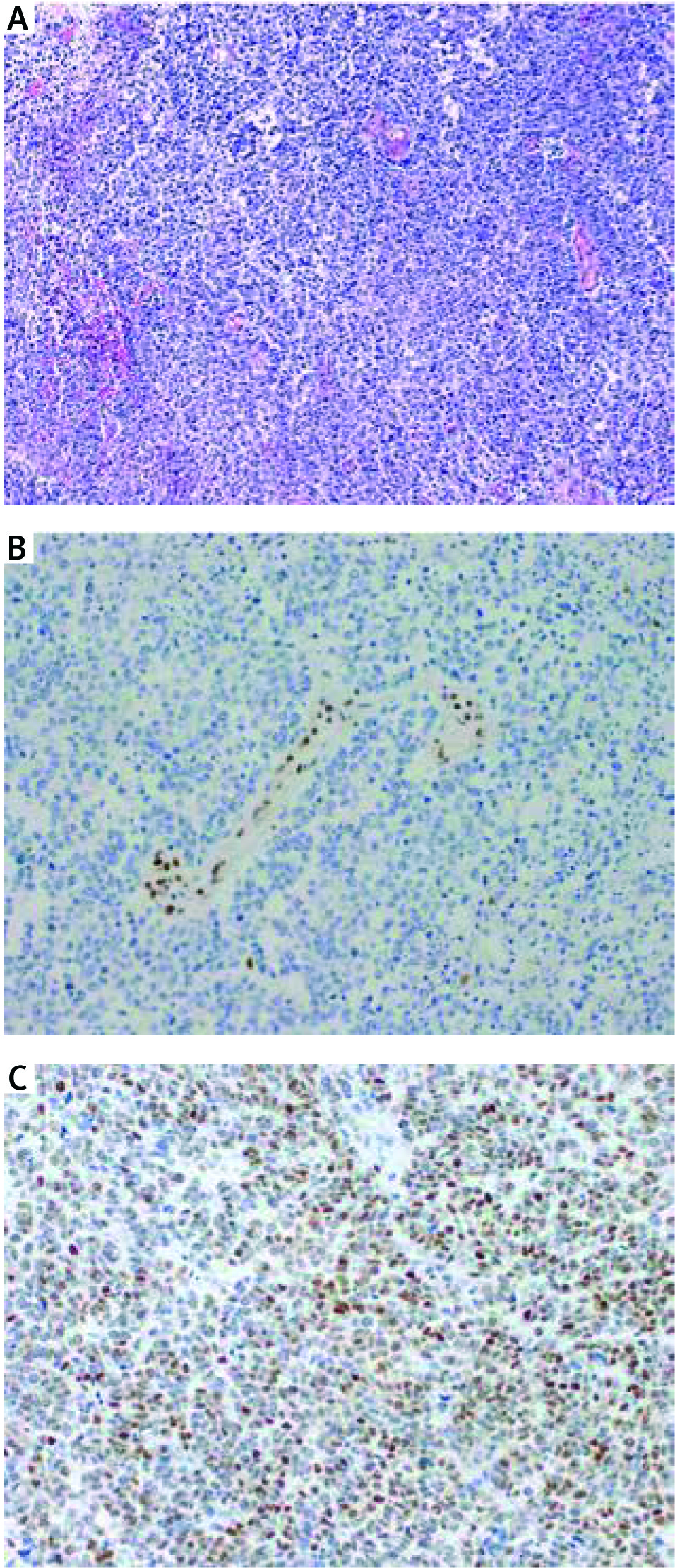
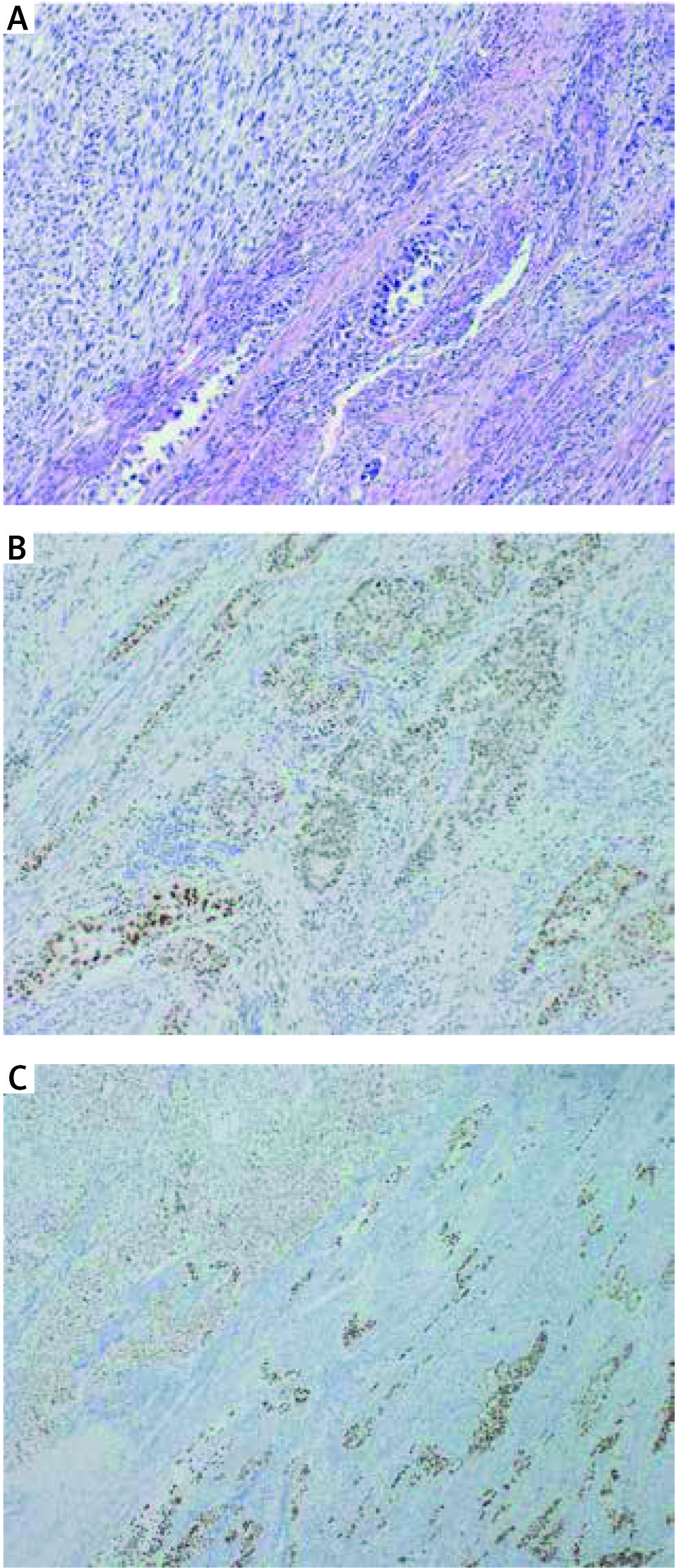
DNA extraction from FFPE tissue sections
Serial 6-μm slices were cut from the paraffin-embedded blocks, and the first section was stained with haematoxylin-eosin (HE) to evaluate the tumour cell content. If the tumour cell content was less than 50%, macrodissection of tumour cell-rich areas was performed. After deparaffinisation, DNA was extracted according to the standard procedure described in the Cobas® DNA Sample Preparation Kit (Roche Molecular Systems, Inc., USA) package insert. The amount of genomic DNA was adjusted to fixed concentrations of 2–5 ng/μl.
TP53 mutation analysis
TP53 mutation status in tissue samples was assessed by PCR using MyTaq HS DNA polymerase (Bioline, Germany) through 37 cycles with appropriate annealing temperatures. Primer sequences and annealing temperatures specific for exons 4–8 of TP53 are shown in Table 1. PCR samples were subjected to direct sequencing of single-stranded PCR products using a BigDye® Terminator v1.1 cycle sequencing kit and an ABI 3500 genetic analyser (Applied Biosystems).
Table 1
Primer sequences specific for TP53 as well as methylated (met) and unmethylated (unmet) MLH1 promoter
MLH1 methylation analysis
Bisulphite conversion of genomic DNA from tissue samples was performed using the InviGene® Bisulfite Conversion Kit (STRATEC Molecular GmbH) according to the manufacturer’s instructions. MLH1 promoter methylation status was assessed by comparing normal genomic DNA with bisulphite-treated DNA of each tissue sample using methylated and unmethylated allele-specific primers and PCR conditions as described above. Primer sequences and annealing temperatures are shown in Table 1. PCR samples were subjected to an Agilent 2100 Bioanalyzer using the Agilent DNA 1000 Kit according to the manufacturer’s instructions (Agilent Technologies).
Statistical analysis
Concordance in immunohistochemical staining between sarcomatous and carcinomatous components was evaluated with Fisher’s exact test and kappa test. Other categorical variables were compared using Fisher’s exact test. Correlation between MMR and p53 status was assessed by Spearman’s rank-correlation test. Calculations were performed using Statistica software (Dell, version 13), licensed to the Medical University of Gdansk.
Results
Twenty patients were included in the study. Immunohistochemical staining patterns of each tumour, along with basic demographic data, are shown in Table 2. Median age at diagnosis was 71.5 years (range 54 to 88, average 72.5). All cases occurred in post-menopausal patients. Immunostaining for hMLH1 and PMS2 were negative in three out of 20 uterine CS (15%). In turn, immunohistochemically, hMSH2 and hMSH6 were present in all tumours. There was a concordance rate of 100% in MMR protein staining between both components (k = 1, p< 0.001).
Table 2
Summary of p53 and MMR alterations in current cohort of uterine carcinosarcoma
We detected abnormal IHC expression of p53 in 12 of 20 samples (60%). In the vast majority of cases (18/20, 90.0%) there was concordance between the sarcomatoid and carcinomatous components (k = 0.895, p< 0.001). Aberrant p53 staining was strong/diffuse in all cases.
Interestingly, we observed that in all three cases with defective MMR, there was no immunohistochemical evidence of any p53 defect; on the other hand, all uterine tumours with defective p53 were positive for MMR proteins. Thus, in the next step, we tested the MLH1 promoter methylation status and TP53 mutations in our cohort (one case was excluded due to the lack of representative material for further analyses). TP53 mutation was detected in 8/19 tumours (42.1%), whereas MLH1 promoter hypermethylation occurred in 4/19 cases (21%). Only one case showed a simultaneous defect in TP53 and hMLH1 (5%). TP53 mutations were found exclusively in regions responsible for DNA binding, and some of them had been previously observed in uterine CS. Agreement between the results of the genetic and immunohistochemical study was moderate for p53 (k = 0.615, p< 0.01) and strong for hMLH1 (k = 0.826, p< 0.01). Correlation between p53 and hMLH1 defect in uterine CS, assessed by Spearman’s rank-correlation, was –0.40849 (p = 0.0825).
Discussion
We hypothesised that loss of mismatch repair (MMR) proteins and p53 expression, caused by genome instability and generation of either microsatellite (MSI) or chromosomal instability (CIN), may play a role in the pathogenesis of uterine CS. It is well known that MMR-deficiency and TP53 mutations, either gain or loss of function, may lead to the development of the sarcomatous component, for example through TGF-bsignalling (by MMR deficiency) [17] or miRNA [18, 19].
In previous studies, conflicting results have been published. In their prototypical study, Taylor et al. examined 28 uterine CS for MSI and MMR protein (hMLH1, hMSH2, and hMSH6) expression and TP53 status [20]. Six tumours (21%) displayed defective DNA mismatch repair owing to the high-level MSI (MSI-H) phenotype in at least one component. Interestingly,immunostaining was negative for hMLH1, hMSH2, and hMSH6 [20] in only two of six cases with MSI-H. Other studies reported IHC MMR protein loss in CSs at various frequencies, ranging from 3% to 40% [21–23]. Only one study reported frequent IHC loss of hMSH2 and hMSH6 in CS [23]. However, the results of the IHC studies of MMR proteins without molecular confirmation should be interpreted with caution. The MSI frequency in CS of the female genital tract ranges from 5% to 23% of cases [20–22, 24, 25]. Overall, the frequency of MMR defects assessed with the use of various methods reported in the literature is 13% [26]. The two main mechanisms of MMR loss in cancer are sequence mutations and epigenetic changes. Extensive parallel sequencing of 22 uterine CSs revealed MLH1 frameshift mutation in one case (5%) and MSH6 nonsense mutation in three cases (15%) [27]. A recent comprehensive epigenetic analysis of 57 uterine CS cases revealed MLH1 epigenetic silencing in two tumours exhibiting MSI, discovering a novel important mechanism responsible for MMR deficiency in uterine CS [28]. Our study reports another four cases of MLH1 promoter methylation in CS, which supports these findings.
We have observed MMR defect in 15% of cases according to the IHC and 21% according to the MLH1 promotor hypermethylation analysis. This discrepancy was shown in a previous study from Australia; in 702 endometrial cancer patients it was demonstrated that 3% of MMR-proficient tumours showed MLH1 promoter methylation, and the methylation levels in these cases were low [29].
Taylor’s cohort and ours showed a tendency towards an inverse association between MMR and p53 status [20]. Only one tumour from our cases presented with MLH1 hypermethylation and TP53 mutation; however, it was still positive immunohistochemically. Moreover, in our cohort, MLH1 promoter hypermethylation occurred preferentially in cases with the endometrioid epithelial component, whereas p53 aberrations were seen in CS with the serous or undifferentiated component. These findings are consistent with another study that demonstrated a higher frequency of p53 defects in CS with serous differentiation when compared with CS with an endometrioid epithelial component [30]. Genetic studies showed that the overall mutation profile of uterine CS corresponds to the mutation profiles of either serous (mutation of TP53 and PPP2R1A) or endometrioid carcinomas (mutation of PTEN and ARID1A) [21, 31]. Nevertheless, these findings are not supported by other studies that failed to demonstrate any inverse associations between mutations in MMR genes and TP53 [21, 28].
Conclusions
We conclude that epigenetic loss of hMLH1 expression may be an element of pathogenesis in some cases of uterine CS. In our cohort, the MMR defect tended to show an inverse correlation with p53 abnormalities. This association may indicate the existence of two distinct pathways of uterine CS development: through microsatellite or chromosomal instability. As CSs are rare tumours, multicentre studies are required to better explain the role of these alternations in tumours originating from the uterus and other organs.






