
Introduction
Oxygen is an indispensable element for most living organisms. Due to its strong reactivity oxygen participates in high energy electron transfers leading to generation of adenosine-5-triphosphate through oxidative phosphorylation. This process ensures the existence and enables the evolution of complex multicellular organisms but also leads to the production of reactive oxygen species (ROS). The term ROS refers to chemically reactive molecules such as superoxide anions, peroxides and hydroxyl radicals that are able to modify DNA and protein structures and change their cellular behavior. First identified as harmful by-products of aerobic metabolism, ROS have been finally recognized as secondary messengers in several intracellular signaling pathways [1, 2].
Reactive oxygen species and melanogenesis
Although ROS are generated mostly in mitochondria and endoplasmic reticulum, they can be produced in other intracellular structures such as peroxisomes and melanosomes [1, 3]. Melanosomes constitute the lysosome-related organelles in melanocytes where melanin synthesis takes place. The melanogenesis pathway diverges in two sub-pathways depending on the final type of melanin: eumelanin or pheomelanin (Fig. 1). Both are initiated by hydroxylation of phenylalanine into L-tyrosine or directly start with L-tyrosine. L-tyrosine is then hydroxylated to L-dihydroxyphenylalanine (L-DOPA) and further oxidized to L-dopaquinone (DQ). These reactions are catalyzed by the first of the three crucial enzymes in the melanogenesis pathway – tyrosinase, a key rate-limiting enzyme of both eumelanin and pheomelanin synthesis. After the formation of DQ, the pathway is divided into two arms leading to the synthesis of either eumelanin or pheomelanin. Pheomelanin synthesis requires the presence of cysteine, which reacts with DQ and forms cysteinyl-DOPA, which is then converted into quinoline and finally polymerized to pheomelanin. Eumelanin synthesis is continued by addition of an amino group to DQ forming dopachrome. Dopachrome can be spontaneously converted to 5,6-dihydroxyindole or enzymatically converted to 5,6-dihydroxyindole-2-carboxylic acid by the other key enzyme of melanogenesis pathway – tyrosinase-related protein 2 (Tyrp2 or DCT). Then, spontaneous polymerization of 5,6-dihydroxyindole and, induced by the third enzyme (Tyrp1) polymerization of 5,6-dihydroxyindole-2-carboxylic acid allows for the final conversion of both compounds to eumelanin [4, 5]. A schematic representation of the melanin synthesis pathway is presented in Figure 1.
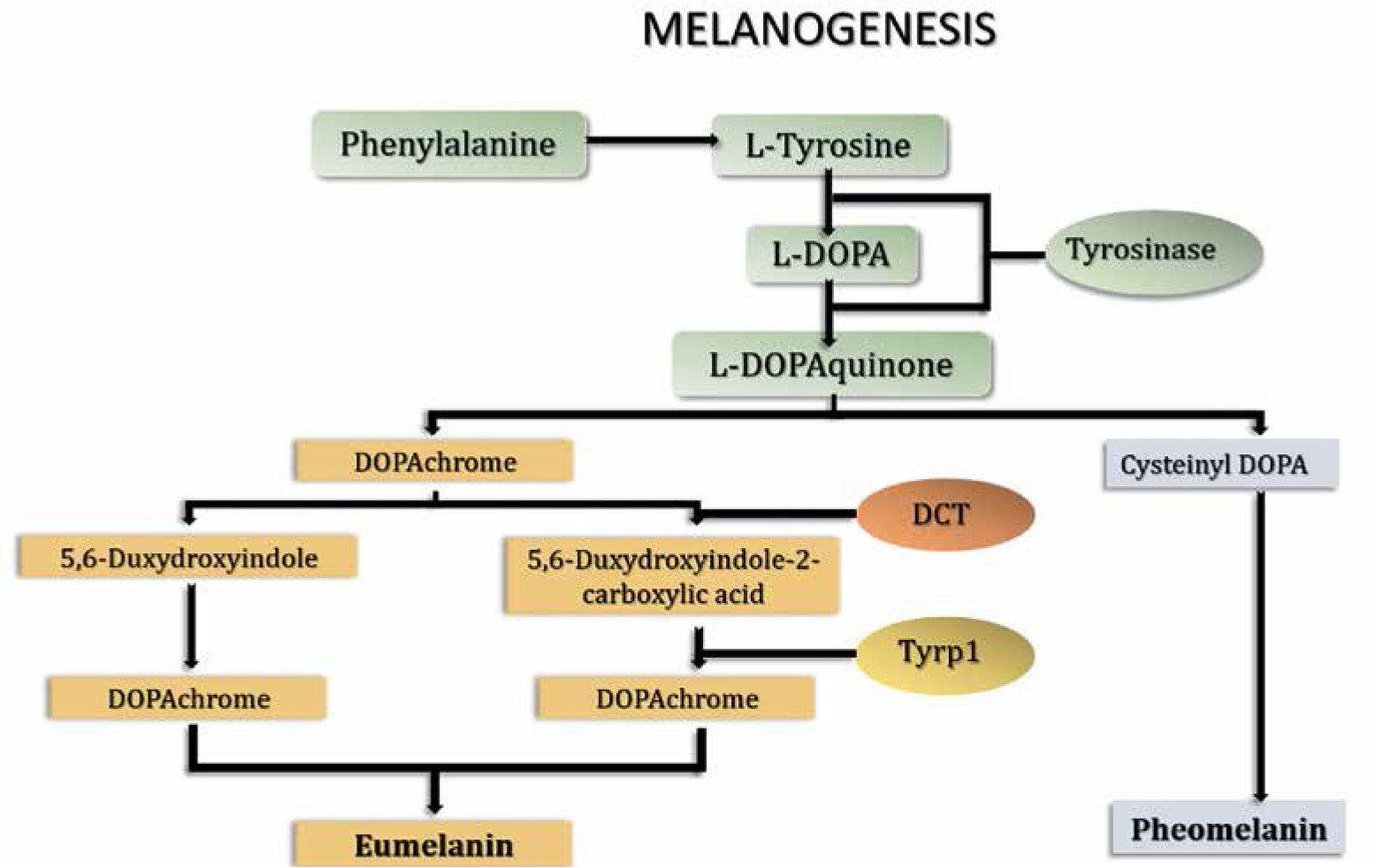
There are several factors that regulate melanogenesis. They include UV radiation, microphthalmia-associated transcription factor (MITF), ERK/MAPK pathway, mitochondrial dynamics, or immune modulation [4]. UV radiation can induce the tumor suppressor protein p53, which regulates the expression and activity of tyrosinase and Tyrp1 [6]. Moreover, UV radiation leads to the increase in ROS levels in keratinocytes and melanocytes, which may cause DNA damage and further promote p53 activation [7]. Microphthalmia-associated transcription factor is a transcription factor which regulates melanocyte development, survival and functions. It also regulates Tyrp1 and Tyrp2. MAPK in turn can indirectly regulate melanogenesis by phosphorylation of MITF, leading to its ubiquitination and degradation, which further results in decreased expression of Tyrp1 and Tyrp2 [8]. Mitochondrial fission causes increased production of ROS, and activates the ROS-ERK signaling pathway, which leads to phosphorylation and proteasomal degradation of MITF and downregulation of melanogenesis [9]. Immune mediators such as cytokines can regulate proliferation and differentiation of melanocytes, as well as melanogenesis. It has been demonstrated that interleukine-4 (IL-4) and interleukine-6 (IL-6) are able to inhibit melanogenesis by suppressing expression of melanogenesis-related genes [10, 11].
Melanin synthesis and oxidative stress in melanoma
The main function of melanin is the protection from photodamage induced by UV irradiation. However, it also regulates the processes which affect melanoma behavior. The pathway responsible for melanin synthesis differs in normal and malignant melanocytes [12]. Skin with higher pheomelanin levels, in comparison to skin with higher eumelanin levels or no melanin, tends to produce more reactive oxygen species, which can promote carcinogenesis [13]. Tyrosinase-related protein 1 has been suggested to increase the eumelanin to pheomelanin ratio and protects against oxidative stress (OS) due to its peroxidase activity [14].
The mechanism and precise impact of melanin on melanoma progression is not clear. Sarna et al. examined the role of melanin pigmentation in melanoma metastasis and found that the presence of melanin inhibits the ability of melanoma cells to spread in vivo [15]. Those results indicate the important role of melanin in melanoma development. Similar results were obtained by Thomas et al., who found a correlation between amelanotic melanoma and poorer survival rates than in pigmented melanomas. However, the lower survival may have resulted from the late diagnosis due to the difficulties with the detection of amelanotic melanoma [16].
Chronic UV exposure affects melanin production by promoting the formation of new blood vessels in the epidermis and tyrosine phosphorylation of vascular endothelial growth factor (VEGFR)-1 and VEGFR-2. Oxidative stress together with hypoxia mediates an increase in VEGFR expression by UVB. Zhu et al. demonstrated that VEGFR activation inhibition leads to decreased tyrosine activity and melanin synthesis [17]. There are also reports describing melanogenesis as a pathogenic factor in melanoma progression. Brozyna et. al. demonstrated that a higher level of melanin shortens overall survival and disease-free survival in patients with metastatic melanomas [18]. Melanin is known to generate reactive oxygen species; thus, melanocytes where the synthesis of melanin takes place maintain higher levels of ROS than other cell types [19]. Pavel et al. found a positive correlation between the levels of melanin and ROS in melanocytes from dysplastic nevi compared to normal skin of the same patient [20]. Since melanoma is a neoplasm originating from melanocytes, melanoma cells are more susceptible to ROS accumulation and exhibit higher levels of ROS in comparison to normal cells. It has been speculated that increased OS by exogenous ROS generation therapy may have a selective effect, killing cancer cells without affecting normal cells [21].
Melanin synthesis appears to act as a double-edged sword: on one hand it protects the cells from UV-induced damage, but on the other hand it leads to higher levels of intracellular ROS, and therefore it can increase melanoma susceptibility [22].
In melanoma, ROS can be additionally generated in mitochondria through the electron transport chain, mainly complex I and III; however, the exact contribution of each complex is not clear [23]. Mitochondrial DNA mutations are rare in cancer; therefore they do not seem to be the main cause of ROS generation and cancer development [24]. It is more likely that the role of mitochondria in cancer is more connected to defective metabolic regulation. Melanoma is a type of cancer that, apart from OS, is especially sensitive to metabolic changes such as the Warburg effect [25]. Melanocytes and melanoma cells feature unique redox regulation and are characterized by their ROS-generating roles. An example of the increased OS effect is red hair pheomelanin, which has a pro-oxidant role and is strongly associated with the increased oxidative DNA and lipid damage [26].
Oxidative stress can alter gene expression by preventing key epigenetic enzymes from interaction with DNA. That in turn may lead to global hypomethylation and genomic instability [27, 28]. Oxidative stress can affect epigenetic modifications in different ways with varied effects. One way of such alterations is the increased promoter methylation due to the reduction of glutathione-s-transferase P1 (GSTP1) expression by induction of the methyl binding protein complex of histone deacetylase (HDAC) and DNA methyltransferase (DNMT) [28]. ROS-induced abnormal DNA methylation pattern modifications are involved in malignant transformation and progression of numerous tumors [29].
Oxidative stress and redox status changes cause the transition from quiescent to proliferative status, growth arrest or even cell death activation depending on the duration and extent of redox imbalance [30]. Reactive oxygen species regulate cell proliferation depending on their amount. High levels of ROS lead to OS and induce apoptosis. Low ROS levels may promote G1 to S phase cell cycle transition [31]. Cell death caused by the high hydrogen peroxide concentration can be inhibited by the antiapoptotic protein Bcl2 with antioxidant activity [32]. This activity of Bcl2 is enhanced by the protein kinases MAPK, ERK1/2, and JNK1, which are activated by OS [33, 34]. Bcl2 antioxidant function is stimulated by its phosphorylation and leads to inhibition of the apoptosis response. Hydrogen peroxide action is a part of the mechanism enabling cell growth stimulation by downregulation of p27, which is an inhibitor of Cdk2 and G1 to S cell cycle transition [35, 36]. H2O2 regulates the cellular localization of p27Kip1 in transformed melanocytes. Addition of hydrogen peroxide to melanoma cells arrested in G1 phase enhances cell proliferation and causes cytoplasmic localization of p27Kip1. It has been suggested that H2O2 scavenging prevents nuclear exportation of p27Kip1, leading to cell cycle arrest. Accordingly, cancer cells can benefit from their prooxidant state by the increased cytoplasmic p27Kip1 localization [37]. There is a strong relationship between ROS and the cell cycle. It was observed that cells treated with antioxidants or deprived of growth factors were characterized by a very low level of ROS and remained in a quiescent state [35]. Overexpression of Bcl2 protooncogene enhances the activity of superoxide dismutase, catalase (CAT) and glutathione peroxidase and decreases ROS production, which in turn inhibits G1 to S transition and reduces cell proliferation. An increased ROS level may not only contribute to genomic instability, but can also be linked with oncogenesis, acting as the mediators of different signaling pathways involved in the enhancement of cell proliferation and tumor formation [31, 38]. Reactive oxygen species level is highly regulated by p53, which induces apoptosis by promoting ROS production; at the same time, p53 induces expression of several antioxidant genes [39, 40]. Also, the enhanced intracellular ROS generation may result from the increased PI3K/AKT signaling or oncogenic Ras expression; such cells are resistant to drug-induced apoptosis [41, 42].
Oxidative stress plays an essential role in melanoma pathophysiology. One of the reasons for the higher ROS levels in melanoma is the production of hydrogen peroxide and consumption of reduced glutathione (GSH) during melanogenesis [30]. Another is that UV radiation exposure, which is a major risk factor for melanomagenesis, causes ROS-mediated OS [43]. Large amounts of ROS contribute to the ability of aggressive cancer cells to mutate, self-renew, inhibit antiproteases, injure local tissues and promote tumor heterogeneity, mutation and metastasis [44]. Cutaneous melanoma develops from epidermal melanocytes in skin, which is a relatively hypoxic tissue. Reactive oxygen species are generated as a result of increased metabolism of transformed cells, immune reaction against tumor, UV radiation, melanin production and an altered antioxidant system [45].
Melanogenesis and oxidative stress correlation in melanoma
The markers of the melanin synthesis pathway are tyrosinase, Tyrp1 and Tyrp2/DCT. The oxidative stress pathway consists of several genes either negatively or positively regulating generation of reactive oxygen species. They include AOX1, NOXO1, BAG2, TTN, SOD1/2/3, GPX8, ENOX1/2, SELENOP, MT3, TTN, GSTM4, SIRT2/3, GSTP1, CYGB, and MSRA. TCGA data analysis of potential gene correlations showed a significant correlation of those markers with genes involved in OS (Fig. 2) [46].
Fig. 2
Heatmap of correlations between three of the gene members of melanogenesis and nineteen genes representing oxidative stress pathway in melanoma patients (n = 444); p-value < 0.05 was considered significant
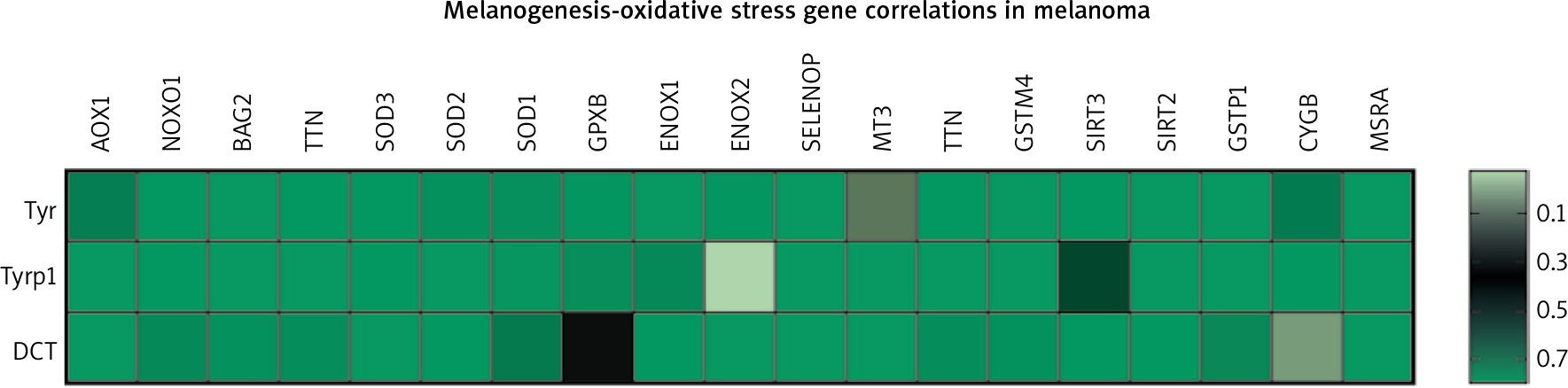
The above data reveal a high level of dependence between melanin synthesis and the production of reactive oxygen species. Thus, manipulating one of those processes can directly influence the other, which should be considered when developing new anti- or pro-oxidant therapeutic strategies.
Reactive oxygen species and markers of oxidative stress
The excessive amount of ROS results in OS, promotion of tumorigenesis and cancer progression. Activation of redox sensitive transcription factors, such as AP-1, p53 and NF-κB, regulates the expression of pro-inflammatory and other cytokines, cell differentiation and apoptosis [47]. At high concentrations ROS may cause serious and irreparable cell damage and lead to apoptosis or necrosis. Overproduction of ROS induces the opening of ion channels, lipid peroxidation, protein modifications and DNA oxidation. Extremely low levels of ROS may in turn result in cell cycle arrest [1]. To ensure cellular homeostasis, the overproduction of ROS needs to be balanced by ROS scavenging. There are two systems responsible for maintaining a low level of ROS: antioxidant enzyme and sulfur reduction buffer systems. The first one contains the superoxide dismutase family enzymes (SOD1, SOD2 and SOD3), CAT and peroxidase. The latter one engages reduced glutathione (GSH), thioredoxin (TRX) and thioredoxin reductase (TRXR). Glutathione peroxidase converts H2O2 to H2O and O2 through transformation of GSH to oxidized glutathione [21].
Biomarkers of OS are the molecules which are modified by interactions with ROS and those of the antioxidant system. A good biomarker for clinical applicability should feature ease of obtaining a biological specimen, stability in various storage conditions and preparation steps, specificity, sensitivity and reproducibility [14]. Oxidative stress biomarkers are important factors in the evaluation of the disease status and potentially health-enhancing effects of antioxidants. The short half-life of ROS makes it difficult to directly measure their concentration. An alternative way of estimating ROS concentration is quantifying the amounts of proteins, lipids or nucleic acids as the OS products [48]. The markers of OS may be divided into organic and inorganic. The two most common organic markers are 8-oxo deoxyguanosine, which is a marker of DNA damage, and malondialdehyde generated during lipid peroxidation. Inorganic markers include excessive levels of iron and copper. Also a high Cu/Zn ratio is a good indication of OS [29]. Lipid peroxidation of polyunsaturated fatty acids (PUFAs) within membrane and low-density lipoproteins (LDL) can be induced by ROS and reactive nitrogen species (RNS). Furthermore, in the case of metabolic syndrome and chronic inflammation, oxidized LDL (oxLDL) drives leukocytes and/or platelets to produce more ROS and RNS [49].
Different organisms have built defensive mechanisms to protect themselves from the toxic effects of excessive ROS and RNS production. This protection acts on three levels: 1) the systems preventing FR (free radical) formation such as inhibitors of enzymes that catalyze FR formation, 2) scavengers and trappers of FR (mainly antioxidants) which eliminate excessive reactivity of ROS by turning them into nonradical and nontoxic metabolites, 3) the final system comes into play when both previous systems fail; the repair mechanisms recognize impaired molecules and decompose them; they include proteinases which degrade oxidatively modified proteins, lipases degrading oxidatively damaged lipids and various DNA repair systems in the case of modified DNA [48].
Oxidative stress and hypoxia
Under normal cellular conditions there is a constant balance between the production of ROS and their elimination by the antioxidant system. Reactive oxygen species generation shares a toxic dependency with hypoxia; both are types of redox stress. In response to hypoxia, proliferative melanoma cells are able to arrest their cycle in G1 phase, and switch their phenotype to become more invasive by altering gene expression [50]. Hypoxia can induce resistance to MAPK pathway inhibitors by mediating upregulation of hepatocyte growth factor/mesenchymal-epithelial transition (HGF/MET) signaling, increasing SNAIL expression and decreasing E-cadherin expression [51, 52]. On the other hand, malignant cells that are distant from blood vessels and experience hypoxia and nutrient starvation have a slow-cycling phenotype. It is especially important since many treatment strategies target only proliferating cells; slow-cycling cells evade drug activity and survive [53]. One of the main players in the cell response to hypoxia is hypoxia inducible factor 1 (HIF-1). It consists of the constitutive HIF-1β and the oxygen-responsive HIF-1α subunits. HIF-1 controls gene expression when the level of oxygen decreases. The elevated expression of HIF-1 and the following increase of transcriptional activity induced by hypoxia have been linked to many tumor types such as head and neck cancers, cervical carcinoma, leukemia, or renal cell carcinoma [54]. In normal healthy cells and in normoxia (21% O2) HIF-1 is hydroxylated by prolyl hydroxylases that allows binding with von Hippel-Lindau protein (VHL), targeting HIF-1 for ubiquitination and at the end its proteasomal degradation [55]. However, during hypoxia, the hydroxylation process is inhibited and the HIF-1α subunit is stabilized. It can translocate into the nucleus and form a complex with HIF-1β. The functional HIF-1 complex binds to the HIF-binding site in hypoxia-responsive elements in the promoter or enhancer regions of its target genes. Active HIF-1 increases anaerobic glucose metabolism and helps cells to survive in hypoxic conditions, but also can promote angiogenesis and tumor invasion [54]. Activity of HIF-1 can also be regulated at the transcriptional and translational levels by PI3K-AKT-mTOR and MAPK/ERK pathways, wherein, under hypoxia mTOR (mammalian target of rapamycin) seems to be inactivated; therefore stimulation of HIF-1α by mTOR is relevant only under mild hypoxia or normoxia [56]. HIF-1 expression may be increased by reactive oxygen species. It has been demonstrated that ROS raise the activity of HIF under normoxia in melanoma cells through transcriptional upregulation of NF-kappaB [57]. HIF-1 cooperates with MITF in the regulation of transcriptional responses to OS. HIF-1 regulates MITF and at the same time acts as its target. Such a feedback loop allows the cell to cope with both hypoxia and OS [58].
Oxidative stress and tumor microenvironment
The tumor microenvironment is composed of several types of cells and extracellular matrix (ECM), including cancer cells, fibroblasts, various types of immune cells, and vascular endothelial cells [59]. It plays a critical role in melanoma invasion and metastatic spread. Normal stroma has the ability to restrain tumor growth, whereas tumor-infiltrated stromal fibroblasts, the so-called carcinoma-associated fibroblasts (CAFs), behave in the opposite manner. Carcinoma-associated fibroblasts are recruited by primary tumor cells and help melanoma cells to escape from the supporting stroma and form metastases. Their action is mediated by secretion of stromal derived factor 1 (SDF-1), vascular endothelial growth factor-A (VEGF-A) and IL-6. Comito et al. investigated the interplay between stromal fibroblasts and hypoxia and its influence on melanoma aggressiveness. They demonstrated that those two components synergistically increase the invasiveness of primary melanoma cells and induce expression of soluble factors such as VEGF-A, SDF-1 and IL-6. The study showed that ROS scavenging destroys HIF-1 accumulation in hypoxia-dependent CAFs and eliminates the expression of IL-6, VEGF-A and SDF-1. That phenomenon suggests a key role of hypoxia driven OS in regulation of the angiogenic and inflammatory response during melanoma progres- sion [60]. Another study describing the role of OS in melanoma immunity confirmed that UV-mediated oxidative damage contributes to inflammation, gene mutation and immunosuppression. Halliday et al. reported that the inflammatory cells such as macrophages and neutrophils produce large amounts of ROS that cause damage to lipids, proteins and DNA, resulting in carcinogenesis [61]. Also the elevated levels of RNS within the tumor microenvironment help to maintain a chronic inflammatory and immune suppressive status, since increased nitric oxide levels can lead to decreased leukocyte proliferation and their decreased infiltration [62].
As mentioned above, melanoma is characterized by extraordinarily high levels of OS in the primary tumor environment, which is due to the structurally aberrant melanosomes of melanoma cells, exogenous attacks, and the infiltration of immune cells [63]. Oxidative stress and inflammation markers have frequently higher expression in melanoma than the surrounding tissues, or other skin cancers. Sander et al. studied superficial spreading melanoma samples and found that CAT, SOD1, and SOD2 levels were significantly higher in melanoma tissue than in age-matched control tissues, nevi, or tumor tissues from squamous cell carcinoma or basal cell carcinoma [64].
Reactive oxygen species may strongly influence the behaviors of different cells within tumor stroma. In tumor-associated macrophages, which are mainly derived from circulating monocytes and are acknowledged as the most abundant leukocytes in melanoma lesions, ROS greatly enhance the secretion of tumor necrosis factor (TNF-α), thereby increasing tumor invasiveness [65]. Reactive oxygen species have been recognized as a strong weapon of the immune system to kill tumors [63, 66]. However, when malignant melanoma cells escape the apoptosis triggered by abundant ROS, persistent ROS may favor melanoma survival, proliferation, and metastasis [63, 67].
Conclusions
Oxidative stress plays an essential role in the transformation and progression of many common cancers, including melanoma. It impacts the tumor microenvironment, the metastatic ability of melanoma cells and their resistance to therapy. Chronic stress greatly enhances susceptibility of melanocytes to oncogenic transformation. Understanding the role of OS in the complex biology of melanoma may reveal the significance and the potential of pharmacological targeting of OS, providing new possibilities of treatment.






