






Introduction
Psoriatic arthritis (PsA) was first described in the nineteenth century and recognized as a unique entity in 1964 [1]. PsA affects peripheral and axial joints, entheses, skin, nails and is associated with many comorbidities, such as premature atherosclerosis, uveitis, cardiovascular and bowel disease. This arthropathy is classified in a heterogeneous group of spondyloarthritis (SpA): separate disorders with overlapping pathogenetic and clinical features. Psoriatic arthritis is a highly heritable polygenic disease. Multiple genes interact with several environmental factors including stress, trauma, infections and trigger an inflammatory response related to innate and acquired immunity in different tissues and organs. Because of heterogeneity of clinical presentation, accurate diagnosis of PsA may be a challenge. Moreover, the lack of effective screening tools as well as not having effective biomarkers result in the underrecognition and delayed diagnosis and treatment, which has serious health consequences, as almost 50% of patients have irreversible joint deformity in the first 2 years of disease onset [2]. The high direct and indirect costs associated with the disease represent a significant economic burden on society [3].
Epidemiology of psoriatic arthritis
There are only a few studies available in the literature regarding the prevalence and incidence of PsA in a population. It is highly variable, influenced by ethnic and geographic variations, different study designs notably case definition and being highest in people of European ethnicity – 0.42% and lowest in Japanese – 0.001% [4, 5]. A population-based study of The Health Improvement Network (THIN) in the United Kingdom which included 4.8 million patients estimated the prevalence of PsA at 0.19 % [6]. In United States study the prevalence was evaluated to be 0.25% [7]. In psoriatic patients the prevalence of PsA ranges wide between 6% and 41%. The manifestation of psoriasis precedes that of arthritis by 10 years on average in 60% of patients, in minority of patients arthritis and psoriasis occur synchronously or psoriatic arthritis precedes the cutaneous-only psoriasis (PsC) [8]. PsA may start at any time of life but the average age of onset is 30–50 years. The frequency is almost equal for both sexes [9].
Pathogenesis of psoriatic arthritis
Genetics of psoriatic arthritis
The role of genetic background in the pathogenesis of psoriatic arthritis is undoubtful. According to twin and family studies performed in European populations, the heritability for chronic plaque psoriasis is estimated to be from 50% to 90% [10–12]. In case of psoriatic arthritis, it is even higher and stands for 80% to 100% [13, 14]. Up to date, multiple large-scale Genome Wide Association Studies (GWASs) have been conducted in chronic plaque psoriasis and led to identification of approximately 70 risk loci. In the GWAS performed by Nair et al. in 2009 on subjects of European ancestry, 1755 cases were known to have psoriasis with PsA and 3523 had psoriasis alone. In this study the authors investigated the association with PsA and the differences between PsA and cutaneous psoriasis. Three loci were associated with PsA when compared to normal controls (HLA-C, IL-12B, and TNIP1). There was a statistically significant difference between PsA and psoriasis alone at three loci (HLA-C, IL-12B and IL-23R). HLA-C and IL-23R were more strongly associated with cutaneous-only psoriasis (PsC), and IL-12B with PsA [15]. LCE3C_LCE3B-del is another known psoriasis risk marker. Noteworthily, the association of SNP correlated with LCE3C_LCE3B-del was confirmed in a population of British patients with psoriatic arthritis, but not in a population of German patients. This discrepancy needs further investigation [16, 17]. Formally, in PsC, only three GWASs based on smaller sample collections have been published so far [18–20]. Together with the candidate loci studies and Immunochip array studies, it led to identification of 13 genetic regions associated with the risk of PsA at significance rate p ≤ 5 × 10-8. They include: SNPs near HLA-B and HLA-C in major histocompatibility complex (MHC ), IL-2, IL-21, IL12B, IL23R, IL23A, TNIP1, TRAF3IP2, CSF2, FBXL19, REL, RUNX3, TYK2, NOS2 and PTPN22. In the context of the studies on candidate genes in patients with PsC, meaning psoriasis without joint involvement for at least 10 years, it is reasonable to consider that PsC presents different genetic architecture than PsA. The main difference is observed within the MHC region in stronger association of HLA-C*06 with PsC and stronger association of HLA-B*27 with PsA [21, 22]. The results of the most up-to-date GWAS including genotyping of 791,217 SNPs in 1,430 PsA case subjects and 1,417 control subjects confirmed previously reported PsA risk regions near HLA-B (rs36058333), IL12B (rs918520), TRAF3IP2 (rs33980500), TNIP1 (rs8177833) and TYK2 (rs35251378) [23]. Further meta-analysis revealed two PsA-specific signals for rs12044149 near IL23R and rs9321623 near TNFAIP3. The PsA-specific variants were independent of previously identified psoriasis variants near IL23R and TNFAIP3 [23]. In 2019 Aterido et al. found a novel PsA SNP at B3GNT2 locus, which presumes the disturbances in glycosaminoglycan metabolism in PsA [24]. Although the identification of new disease-specific genetic variants is challenging, there is a high need for further GWASs in PsA.
Table 1 presents non-MHC psoriatic arthritis genetic risk markers identified by GWAS [25].
Table 1
Non-MHC psoriatic arthritis genetic risk markers identified by GWASs [25]
Immunopathogenesis of psoriatic arthritis
Genetic predisposition together with environmental triggers, in which gut dysbiosis favors the ingenerating tissue damage by damage-associated-molecular-patterns (DAMPs), induce release of pathogen-associated-molecular-patterns (PAMPs), or repetitive entheseal mechanical stress production of inflammatory infiltrates consisting of monocytes, dendritic cells (DCs), other antigen presenting cells (APCs), neutrophils as well as T cells in the enthesis and synovium [26, 27] (Figure 1). Pathogenesis of psoriatic arthritis (PsA) starts with activation of the innate immune system cells, i.e. the natural killer (NK) cells, DCs, gamma/delta T-cells, innate lymphoid cells (ILC), polymorphonuclear neutrophils (PMNs) and monocytes/macrophages, and next, the adaptive immunity cells, i.e. the CD8+ T-cells, CD4+ Th1, Th17, Th9 and Th22, become activated, which is bound to intensify the inflammatory response in the joint [28]. In PsA, the Toll-like receptor (TLR)-2 and TLR-4, the cluster of differentiation 40 (CD40), nicotinamide adenine dinucleotide phosphate oxidase (NOX)-2 and cathelicidin LL-37 (leucine-leucine-37), which handle antimicrobial and immunomodulatory functions, exhibit increased expressions [26, 29]. Following recognition of any danger signals by the pattern-recognition receptors (PRRs), the DCs release antimicrobial peptides as well as chemokines and cytokines, among others: IL-12 and IL-23, which leads to differentiation of the naïve T cells into Th1 and Th17 cells, respectively, and to a decreased T regulatory (Treg) cells’ production [28]. The abnormal activation of the IL-23/Th17 axis is the dominant pathology in PsA. IL-23 regulates the Th17 cells by inducing signaling pathways of tyrosine kinase 2 (Tyk2), Janus kinase 2 (JAK2) as well as the signal transducer and activator of transcription 3 (STAT3) which together stimulate the production of IL-17, IL-21, IL-22, G-CSF and tumor necrosis factor α (TNF-α), thereby favoring tissue inflammation. Apart from IL-23, the differentiation of the naïve T cells into Th17 cells is also stimulated by IL-1β, IL-6 and by the transforming growth factor β (TGF-β). IL-17 induces intracellular cascades containing the TNF receptor associated factor (TRAF) 2, TRAF5, TRAF6, mitogen-activated protein kinase 1 (MAPK1) or the nuclear factor-κB (NF-κB). IL-17 is necessary but insufficient for PsA to develop since an involvement of IL-23 as a mediator in Th17 cells’ activation is indispensable [26, 28, 30].
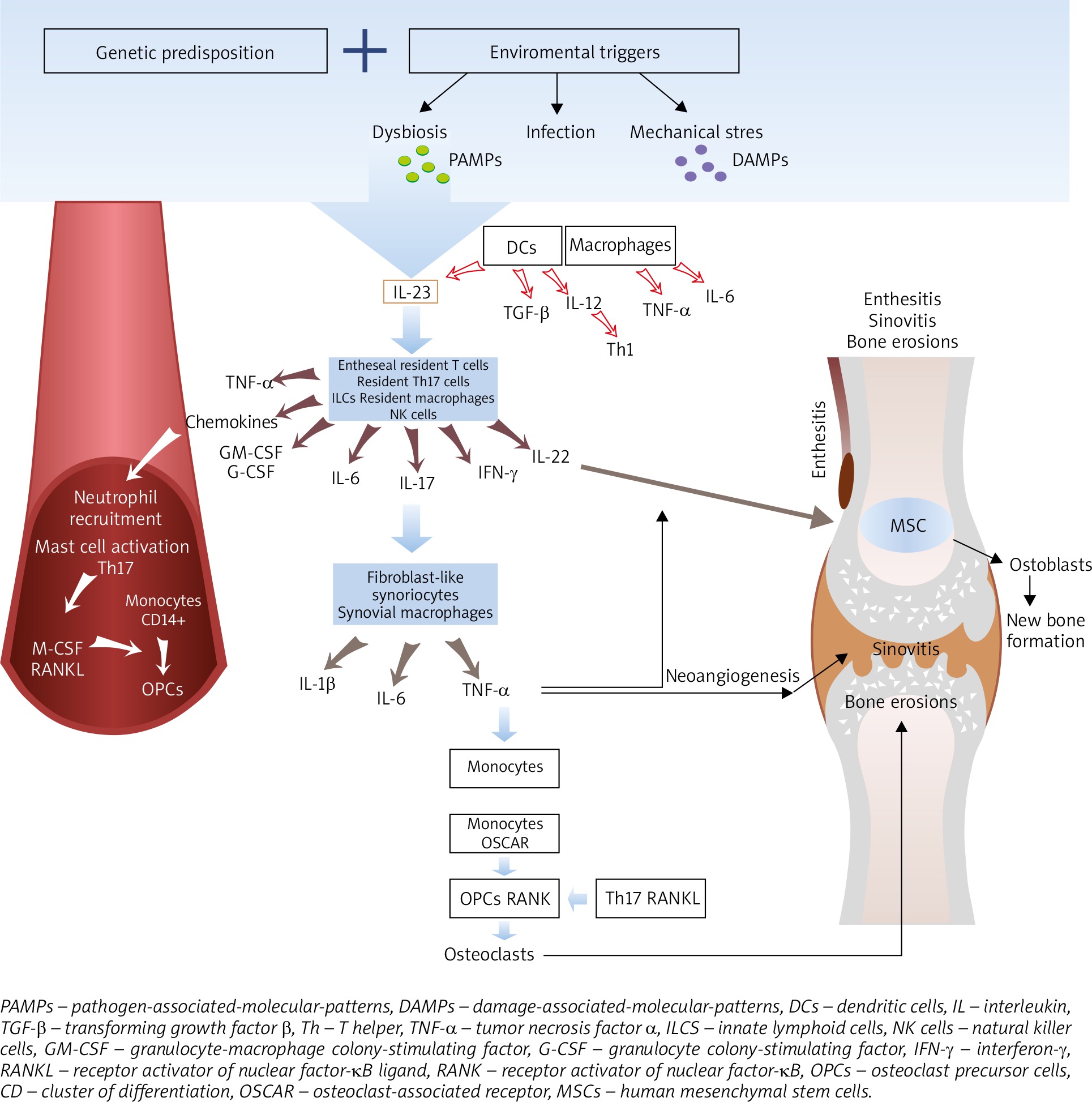
In PsA, apart from the IL-23/IL-17 cytokine axis, the TNF-α is an important effector cytokine in PMNs and mononuclear cells’ recruitment and activation, osteoclasts stimulation as well as production of proinflammatory cytokines leading to synovitis, neoangiogenesis, bone resorption and cartilage destruction. The TNF-α interacts with the TNF receptors (TNFRs) and activates transcriptional factors, such as the NF-κB, activator protein (AP)-1 or AP-2 [26, 31].
In PsA, inflammation may affect the synovium, tendons, entheses, bursae as well as the cartilages and bones [32, 33]. The enthesis is thought to be the initial site of the inflammatory process which spreads to other periarticular and articular structures leading to the development of synovitis, dactylitis, spondylitis, osteitis [32]. Normally, the enthesis has no blood vessels, in enthesitis, however, vascularization is observed. An important early mediator of enthesitis is prostaglandin E2 (PGE2) which evokes vasodilatation and facilitates neutrophils’ recruitment from the bone marrow into the entheses. Neutrophils augment inflammation by releasing proteases and reactive oxygen species (ROS). PGE2 also promotes production of IL-17 by T-cells [34]. In addition, the enthesis contains a specific immune microenvironment which can be activated by mechanical stress and microbial factors [35]. Entheseal resident macrophages, NK cells and T cells, retinoic acid-related orphan receptor-gamma t(ROR-γt+) CD3+CD4-CD8-IL-23R+ T cells are activated by IL-23 produced by DCs [36]. The entheseal resident cells and other innate immune cells are regarded as the main sources of IL-6, IL-17, IL-22 and chemokines (C-X-C motif ligand 1) [27, 30]. The resident γδ T cells and type 3 innate lymphoid cells (ILCs) release cytokines, i.e. TNF and IL-17, which increase PMNs infiltration. What is more, close proximity between the enthesis and perientheseal bone marrow allows recruitment of inflammatory cells from the bone marrow, thereby contributing to osteitis with formation of bone erosions [26, 32, 36]. In PsA, the bone marrow edema and cellular infiltration by the T cells (CD4+, CD8+) andCD68+ macrophages as well as proliferating fibroblasts are present [36, 37].
Progression of inflammation from the extra-articular structures, i.e. extensor tendon enthesitis and peritendon inflammation, extends to the intra-articular inflammation assuming the form of synovitis [32]. PsA synovitis is characterized by lining-layer hyperplasia with an increased number of fibroblast-like synoviocytes and macrophages, tortuous hypervascularity, the presence of hyperemic villi and a subsynovial infiltrate of the T cells, B cells, neutrophils, mast cells and monocytes/macrophages. Elongated, dilated, bushy and tortuous vessels are suggestive of increased angiogenesis [36, 38]. Pro-angiogenic factors, i.e. vascular endothelial growth factor (VEGF), TGF-β, angiopoietin 2, are present in the synovial fluid in the course of PsA [36, 38]. The T cells in the synovial fluid include both CD4+ IL-17+ and CD8+ IL-17+, with the predominance of the latter [36]. The level of CD8+IL17+ correlates with the activity and progression of the articular changes in PsA [39]. CD8+IL-17+ T cells in the synovial fluid are the main source of IL-17, IL-2, IL-6, interferon-γ (IFN-γ) and IL-22 [33, 40]. Thus, the synovium in PsA has a high expression of IL-17A as well as of the IL-17 receptors, where the IL-17A cytokine is directly involved in the osteochondral destruction. The serum IL-17 concentration remains unchanged in PsA, the number of IL-17-producing cells, however, is increased [26, 30].
Bone remodeling, not limited to bone destruction but characterized by subsequent new bone formation, is a unique feature of PsA [30]. In PsA, IL-17 increases osteoclastogenesis, whereas IL-22 promotes new bone formation. Thus, a proper balance between IL-17, IL-22, and IL-23 is essential for maintaining the bone homeostasis [41]. The striking features of PsA are subchondral perientheseal edema and diffuse bone edema [36]. The subenthesis bone in PsA represents increased vascularity and hyperosteoclastic cystic and erosive changes [42]. Physiologically, bone homeostasis is maintained by the balance between osteoclasts, capable of bone resorption, and osteoblasts, responsible for bone formation. In the systemic inflammatory state, stimulation of the CD14+ monocytes by the macrophage colony-stimulating factor (M-CSF), TNF-α and receptor activator of nuclear factor-κB ligand (RANKL) results in formation of osteoclasts’ precursors (OCPs) in the peripheral blood. Additionally, IL-23 and IL-17 independently induce myeloid cells to undergo osteoclast formation [43]. In the subchondral bone, joining of the receptor activator of nuclear factor-κB (RANK), present on the surface of monocytes/macrophages, with RANKL, a membrane protein present on the surface of Th17 cells, stimulates differentiation of monocytes/macrophages into osteoclasts. The activated osteoclasts start to secrete bone matrix degradation enzymes, i.e. tartrate resistant acid phosphatase (TRAP), matrix metalloproteinase 9 (MMP9) and cathepsin K (CatK) which resorb the mineralized bone. Moreover, on the surface of monocytes the osteoclast-associated receptor (OSCAR), following induction by TNF-α, potentiates the actions of RANKL, thereby increasing osteoclastogenesis. RANKL also increases activation of the T cells and their production of pro-inflammatory cytokines. Many cytokines, including TNF-α, IL-1β, IL-6, IL-15, IL-17, IL-23, VEGF, stimulate RANKL expression leading to erosion at the bone-pannus junction and subchondral bone [36, 37, 43].
In PsA, IL-22 promotes the proliferation of the human mesenchymal stem cells (MSCs) and their differentiation into osteoblasts [30]. It induces osteoproliferation in the enthesis and periosteum via the STAT3 activation on osteoblasts causing new bone formation manifested by the peripheral joint fusion, enthesophytes, spurs, ankylosis as well as syndesmophytes in the axial skeleton and changes in the sacroiliac joints [28, 42, 41]. Mechanisms responsible for new bone formation in PsA may result from activation of several signaling pathways, including Wnt (Wingless-type like signaling), BMP (bone morphogenetic protein) and the Hedgehog signaling pathway. BMP promotes osteoblast formation by stimulating expression of the osteogenic transcription factors, such as RUNX2 and osterix [30]. The expression of BMP is induced by the IL-1β and TNF-α [44]. On the other hand, IL-17A inhibits osteoblast differentiation, inducing the expression of secreted Frizzled-Related Protein 1 which is an inhibitor of the Wnt/β-catenin pathway. Similarly, TNF-α stimulates production of the Wnt/β-catenin pathway inhibitors, i.e. sclerostin and Dickkopf-related protein 1 (DKK-1), which are negative regulators of the bone mass. Osteoprotegerin (OPG), a crucial inhibitor of osteoclastogenesis, is a decoy receptor for RANK that prevents its binding with RANKL and, therefore, OPG participates in new bone formation [30, 45, 46].
Pro-inflammatory cytokines secreted in the course of psoriasis may stimulate chondrocytes to produce destructive proteases, which leads to the loss of proteoglycans, impairment of collagen bundles with concomitant release of cartilage oligomeric matrix protein (COMP). COMP, a glycoprotein belonging to the thrombospondin family, is one of the components of the extracellular joint-cartilage matrix. It is believed that an increased level of COMP in the synovial fluid and serum advocates remodeling and cartilage repair processes. Narrowing of the joint space reflects the thinning of the articular cartilage [46, 47]. An increased COMP expression is found not only in the cartilage but also in the tendons, entheses and ligaments. Cartilage destruction in PsA is connected with upregulation of the TNF-α and subsequent increased production of matrix metalloproteinase (MMP) from macrophages [33].
Clinical presentation of psoriatic arthritis
John Moll and Verna Wright as first provided evidence from epidemiological, clinical, serological and radiological studies which confirmed the association between psoriasis and a specific form of arthritis [48]. The authors defined the disease as an inflammatory arthritis occurring in the presence of psoriasis, usually in an absence of the rheumatoid factor in the serological test. They distinguished 5 clinical subtypes categorized by localization of joint involvement: asymmetrical oligoarthritis with less than 5 joints affected, symmetrical polyarthritis affecting 5 or more joints, axial disease, resembling ankylosing spondylitis but with typical asymmetric inflammation of the sacroiliac joint, arthritis mutilans – extreme severe disabling and disfiguring variant with marked bone resorption or osteolysis in hands and feet and distal interphalangeal joint arthritis (DIP). The last pattern, which affects distal interphalangeal joints of the hands, feet, or both, usually coexists with other subtypes. The Moll and Wright criteria were proved to be sensitive, except early forms of PsA, but without being specific. Patterns may pass one in each other over time, specially oligoarthritis into polyarthritis and change along with disease modifying treatments. Each pattern may have overlapping features with other rheumatic diseases: rheumatoid arthritis in symmetrical polyarticular diseases, reactive arthritis in oligoarticular pattern, ankylosing spondylitis in axial form and osteoarthritis in distal interphalangeal joint involvement.
As specific PsA treatments were introduced in the early 2000s, proper classification has become a high need. In 2006, the Classification Criteria for Psoriatic Arthritis (CASPAR criteria) with a specificity of 98.7% and a sensitivity of 91.4% allowed to define psoriatic arthritis for the purpose of enrolling patients into clinical trials and provide guidance to clinicians in patients with long-standing diseases [49]. The CASPAR criteria have contributed to increase awareness of PsA, its early diagnosis and they facilitate cooperation between rheumatologists and dermatologists. In early PsA usefulness of CASPAR criteria is lower, although some studies proved their usefulness also in patients with early disease [50]. According to CASPAR criteria, psoriatic arthritis is diagnosed in a patient with inflammatory musculoskeletal disease: joint, peripheral, spine or entheseal and with at least 3 points from the criteria presented in Table 2 [49]. From these points the current and past psoriasis is a key criterion. Of note, without inflammatory arthritis which is the entry criterion the other criteria of CASPAR should not be considered in diagnosis of PsA.
Table 2
The CASPAR criteria for Psoriatic Arthritis. Modified from [50]. A patient must have inflammatory articular disease (joint, spine, or entheseal) with three or more points from the following five categories
The most common pattern of PsA is the peripheral arthritis which varies from asymmetric oligoarthritis to symmetric polyarthritis. The pattern of joint involvement itself may also change during an individual’s disease course. Peripheral arthritis may be accompanied by axial disease. Peripheral or axial morning stiffness lasting more than 30 min, impaired mobility and fatigue, back or buttock pain are common features of PsA. Synovitis presents clinically as joints tenderness and swelling. Enthesitis, inflammation of the connective tissue between bone and tendon or ligament, is a common manifestation, occurring in up to 60% of patients with PsA [51]. Some authors hypothesize that PsA, contrary to rheumatoid arthritis, is an entheseal-driven disease with enthesitis playing the primary role with a secondary spread of inflammation in the synovium [52]. As imaging studies have shown enthesitis precedes clinical signs and symptoms of PsA for a long time with the exposure to microtrauma and stress of predisposed individuals and may be asymptomatic at the early stage of PsA [53]. Patients may have enthesitis as the only presentation of PsA without evidence of arthritis. Gisondi et al. have demonstrated in ultrasonography (US) of entheses that enthesitis is more common than it is clinically evident and affects more often patients with psoriasis than healthy population [54]. Enthesitis may affect the Achilles tendon, plantar fascia, greater trochanter patella, iliac crest, epicondyles and supraspinatus insertions but other tendons may also be involved. Enthesitis in the distal interphalangeal joints is associated with psoriatic nail plate dystrophy [53]. Lower extremities are more frequently involved. Tenderness over entheses rarely accompanied by swelling may cause intense pain. The severity of enthesitis is thought to be a marker of proliferative and erosive features, such as joint ankylosis, arthritis mutilans and periostitis [32]. Dactylitis (sausage digit) is the second hallmark of PsA that affects 16–48% patients at some point of their disease and classically presents as the association of distal and proximal interphalangeal arthritis with flexor tenosynovitis and enthesitis affecting more often a toe than a finger [55, 56]. Inflammation of the entire finger may have an acute course with swelling, pain, redness of the finger or chronic presenting with swelling but without inflammation [57].
In 85% of patients, cutaneous psoriasis precedes arthritis for about 10 years, putting dermatologists in a “privileged position” to recognize psoriatic arthritis early. On the other hand, as many as 15% of psoriasis patients have undiagnosed arthritis, which has serious consequences for the patient long-term outcome [58]. Patients seen in the clinic within 2 years of diagnosis had less damage and progression than those seen later in their disease course. Another study showed that even a delay of 6 months in consultation led to more severe disease and the formation of erosions, sacroiliitis and worse HAQ scores [36, 59].
The crucial element of PsA diagnosis is clinical evaluation including a combination of history, clinical exams, laboratory findings and imaging methods. Some data in the medical history taken from the patient may be supportive to identify PsA in a patient with cutaneous psoriasis and joint pain. They include positive family history of psoriatic arthritis in first-degree relatives as well as special localizations of psoriasis [9, 56, 60, 61]. Nail involvement including pits and onycholysis is observed in up to 80% in patients with PsA vs. 40% in patients with cutaneous psoriasis. Common involvement of nails in PsA is associated with arthritis of the distal interphalangeal joint and inflammation of the nail bed, which may represent enthesitis as the nail bed and interphalangeal joint have common tendinous insertions [60, 61]. The other localizations that increase the PsA risk in psoriatic patients include scalp psoriasis (2.35-fold increased risk) and the anogenital area involvement (3.98-fold increase) [9]. In addition, it has been shown in a German large cohort study that the risk of arthritis increases with the severity of psoriasis – more than 3 body sites affected by psoriatic skin increase the risk with 2.24 times [62].
Unfortunately, as of 2020, there are no biomarkers that facilitate the early diagnosis of psoriatic arthritis in patients with psoriasis available. There are no laboratory tests which help to make a definite diagnosis. Approximately 25% of patients with psoriatic arthritis are HLA-B27–positive, more common in the axial subtype of disease. Increases in nonspecific indications of inflammation, the serum C-reactive protein level, the erythrocyte sedimentation rate, or both are observed in only 1/3 of patients [63]. Different imaging methods are available for PsA diagnosis. In plain radiography the occurrence of bone and cartilage destruction with pathologic new bone formation is one of the most distinctive aspects of psoriatic arthritis but early inflammatory changes in PsA affect soft tissues and cannot be detected with this method. Ultrasound (US) plays an important role among imaging methods especially in identifying early forms of PsA. This is a low-cost, highly sensitive and non-invasive imaging technique recommended to estimate synovitis within joints, dactylitis as well as enthesitis. US allows for early recognition of anatomical changes, detecting subclinical changes, guidance for local treatment and therapy monitoring [53]. Magnetic resonance imaging (MRI) is the most important tool in detecting structural damage in early PsA. The method is particularly useful in identifying sacroiliitis, seen even years before changes are seen on plain radiography [64].
Current and emerging strategies of psoriatic arthritis treatment
The delayed diagnosis of PsA and, therefore, deferred launching of its treatment, may result in the development of extra- and intraarticular changes even before the first clinical signs are observed. Therefore, early pharmacological intervention, recommended by the European League Against Rheumatism (EULAR), is likely to prevent irreversible bone and cartilage changes [65, 66].
NSAIDs, especially naproxen and COX-2 inhibitors, due to their lower cardiovascular and gastrointestinal side effects, are the first-line treatment of PsA with musculoskeletal manifestations [65–67]. They are fairly effective in the treatment of axial and peripheral enthesitis, however, in active PsA, i.e. arthritis/synovitis in multiple joints (≥ 5), structural joints radiographic changes, with relevant extra-articular manifestations and increased blood inflammatory markers or dactylitis, their use exceeding 3 months is not recommended [66, 67]. When these signs are present, a conventional synthetic disease-modifying antirheumatic drug (csDMARD) must be started promptly. Methotrexate is the first treatment of choice, especially in PsA patients with the skin involvement. The use of other csDMARDs, including sulphasalazine, leflunomide and cyclosporine A, is considered in the patients in whom methotrexate is contraindicated. In PsA patients with the axial involvement or severe enthesitis, as well as in those in whom the csDMARDs are ineffective, biological DMARDs (bDMARDs) or targeted synthetic DMARDs (tsDMARDs) should be applied [26, 66, 67].
Better understanding of genetic, epigenetic and immunological factors involved in the PsA pathogenesis is contributing to elaboration of specifically targeted therapies. The introduction of bDMARDs and tsDMARDs in PsA has substantially improved the functional status, disease prognosis and patients’ quality of life. Since the TNF-α is a key cytokine in the PsA pathogenesis, its antagonists have been proven to be effective in peripheral arthritis, enthesitis, dactylitis and axial arthritis. Five biological drugs, i.e. infliximab, etanercept, adalimumab, certolizumab pegol, golimumab as well as their biosimilars counteracting TNF-α, are the first-line bDMARDs used in the treatment of PsA [41]. The TNF-α inhibitors may reduce the infiltration of T cells and macrophages in the synovium, they are also capable of decreasing the Th17 level in the synovial fluid and then cause theTh17 outflow from the synovium into the lymph nodes and bloodstream. They also reduce the level of circulating OCPs in the patients with erosive PsA [27, 37]. In the developed PsA, the TNF inhibitors slow down the progression of structural joint damage and reduce bone resorption, but they do not prevent new bone formation [41].
Recognition of the essential role of the IL-23/Th17 axis in the PsA pathogenesis has led to the development of biological drugs targeting IL-23 and IL-17. Ustekinumab, a monoclonal antibody against p40 subunit of IL-12 and IL-23, has already proved to be effective in the treatment of PsA with symptomatic and radiographic improvement. Inhibition of IL-12/IL-23 in PsA is effective in peripheral arthritis, enthesitis, dactylitis as well as in the nail changes [41, 43].
Guselkumab, tildrakizumab and risankizumab are three monoclonal antibodies targeting the p19 subunit of IL-23 which are in Phase III of Randomized Controlled Trials (RCTs) and awaiting approval to be used in PsA. According to the results of Phase II Trials, the PsA patients who underwent treatment with risankizumab showed improvement both in the joint and skin symptoms as well as inhibition of radiographic progression [26, 30, 42].
The use of secukinumab, an anti-IL-17 agent available for peripheral PsA treatment, results in the vast improvement of the patient’s physical functioning and their quality of life. Secukinumab is effective in dactylitis, enthesitis and in preventing radiologic progression. It prevents narrowing the joint space and bone erosions, however, there are no data providing evidence on its effect on new bone formation. Ixekizumab, a humanized IgG4 antibody binding IL-17A, is effective in the peripheral joint symptoms, dactylitis and structural damage but it is less effective in the entheseal manifestations [30]. Brodalumab, an anti-IL-17 receptor A, favors improvement in enthesitis, dactylitis and prevents radiographic progression. As IL-17F has an important role in the chronic tissue inflammation, bimekizumab, which is a monoclonal antibody capable of neutralizing both IL-17A and IL-17F, is a very promising drug in the management of inflammatory arthritis [30, 42, 68].
csDMARDs and bDMARDs are effective in the majority of PsA patients, however, there is still a group unresponsive to this type of treatment. Therefore, introducing the drugs which modulate pro-inflammatory intracellular pathways, such as intracellular enzyme phosphodiesterase (PDE), the transcription factors Janus kinase (JAK) and signal transducer of activators of transcription (STAT), may be a good option. Apremilast is an oral, synthetic small molecule that inhibits the phosphodiesterase 4 (PDE4) and subsequently decreases the expression of IL-2, TNF-α, IL-12 IL-17, IL-23, simultaneously increasing the synthesis of IL-10. Apremilast is associated with moderate improvements in enthesitis, dactylitis, physical function and quality of life. Identification of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) axis that regulates the cytokine signaling has contributed to the development of oral inhibitors of the intracellular tyrosine kinases. The JAK inhibitors prevent the activity of one or more JAKs (JAK-1, JAK-2, JAK-3, Tyk-2). Tofacitinib, an oral inhibitor of JAK3, JAK1 and JAK2, inhibits the STAT3 and STAT1 expression, thereby inhibiting the IL-2, IL-4, IL-6, IL-12, IL-15, IL-21, IL-23 and IFN-γ production. Since the cytokines are involved in enthesitis, synovial inflammation, bone resorption, the JAK inhibitors are bound to be effective in PsA. Other tsDMARDs targeting the JAK/STAT signaling are under evaluation in the clinical trials (Table 3). The results of Phase II Trial (EQUATOR) prove the efficacy and good tolerability of filgotinib in PsA, while the results of the SELECT-PsA study show significant improvement in the joint symptoms of PsA patients treated with upadacitinib versus placebo [30, 69, 70]. Intracellular signaling pathways of osteoclasts are promising novel therapeutic targets in PsA [30].
Table 3
Biological and targeted synthetic DMARDs under experimental trials and in the preregistration phase for psoriatic arthritis
Conclusions
Studies in the last decades using new genomic study techniques like GWAS and immunochip have allowed to identify many associated genes which interact with environmental factors and contribute to complex pathogenesis of psoriatic arthritis. As joints damage may start at the initial phase of the disease it has become evident that patients need to be identified and treated early in order to avoid the poor clinical outcome. Hopefully progress in research of psoriatic arthritis pathogenesis will result in advances in the management of the disease and introduction of new therapeutic options, which give hope for a better disease long-term prognosis.