
Introduction
Pituitary stalk thickening (PST) is a relatively rare abnormality in children. The diagnosis may be challenging due to its diverse clinical picture.
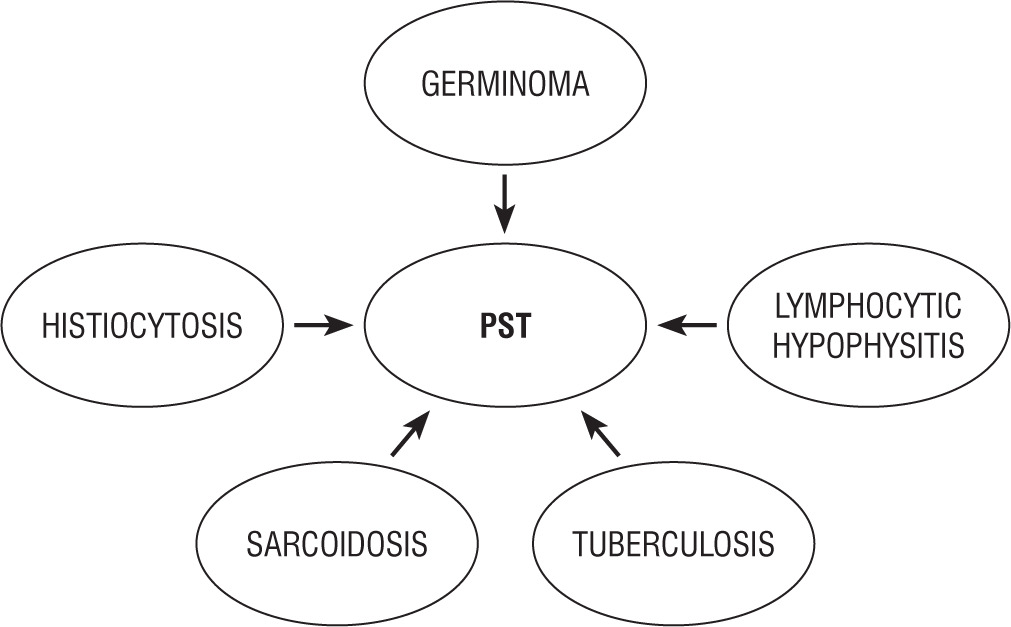
PST develops for a variety of reasons, such as neoplastic, inflammatory, infectious diseases, or congenital abnormalities (Figs. 1, 2). PST mainly affects teenagers and is often detected with magnetic resonance imaging (MRI) in patients with central diabetes insipidus (CDI) [1]. Sometimes, pituitary stalk lesions are accidentally detected during brain MRI. In most cases, the diagnosis is based on radiological imaging and laboratory tests. Because of the risks associated with obtaining a pituitary stalk biopsy, only a small subset of patients had their diagnoses histopathologically confirmed [2]. Despite the development of MRI imaging techniques, PST remains a conundrum for clinicians.
Figure 1
T1-weighted images, sagittal plane. A) 10-year-old boy with germinoma involving pituitary stalk. Pituitary stalk thickening (9 × 8 mm). B) 2-year-old girl with multifocal histiocytosis with involvement of hypothalamic-pituitary region. Pituitary stalk thickening (3.5 mm). C) 16-year-old girl with lymphocytic hypophysitis. Pituitary stalk thickening (4 mm), enlargement of the pituitary gland (11 × 14 × 10 mm), in the intermediate lobe hypointense lesion (8 × 4 × 5.5 mm) – pituitary cyst. D) 5-year-old boy with hyperintense congenital lesion in infundibular recess (3 × 2 mm) – ectopic posterior pituitary
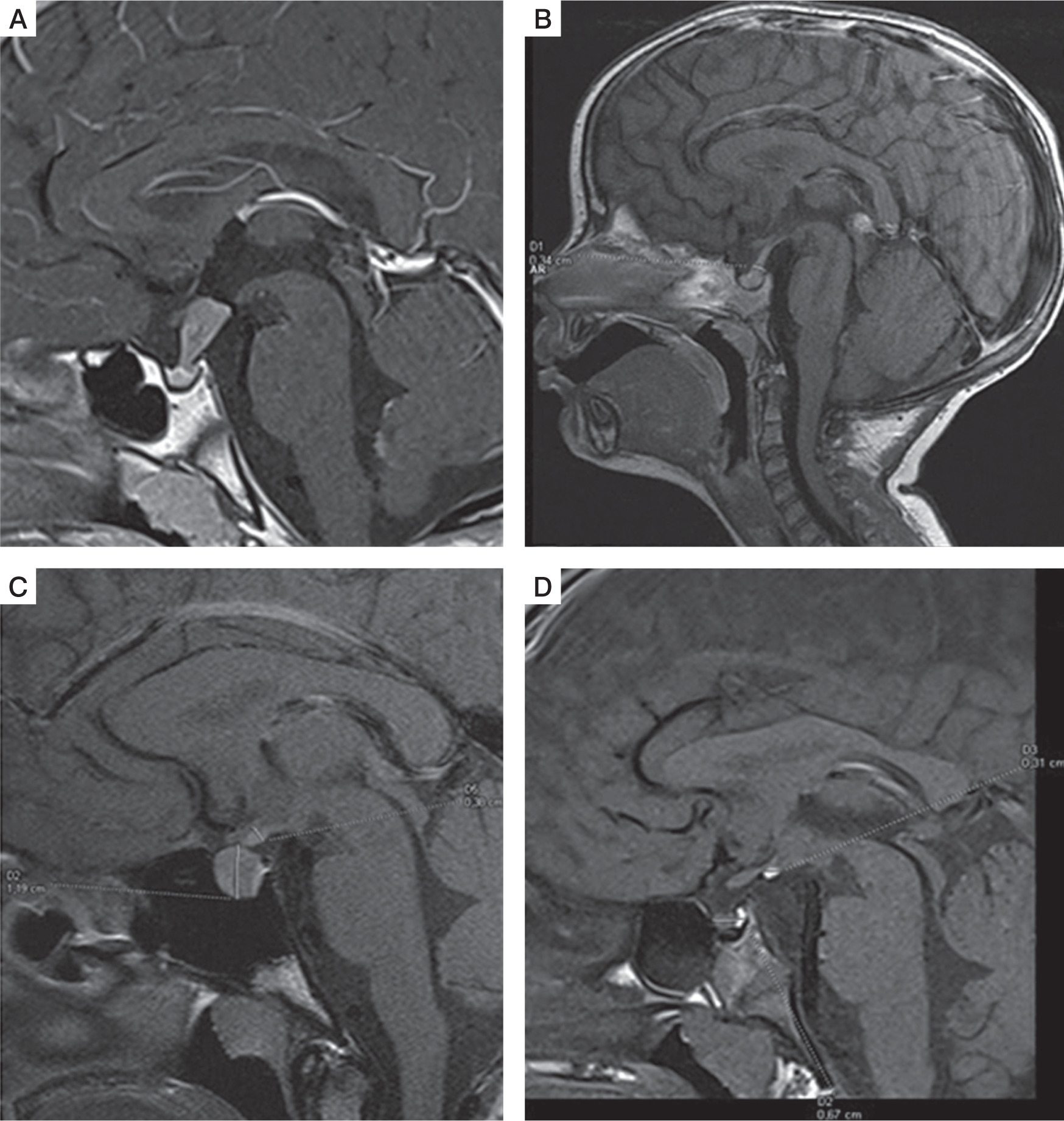
Previous studies determined the correct pituitary stalk dimension as 3.25 ±0.56 mm in the transverse diameter at the optic chiasm and 1.91 ±0.40 mm at its pituitary insertion in the adult population [3]. Godano et al. [4], in the only paediatric study involving 102 children aged 7–12 years, reported smaller stalk sizes (2.35-2.82 mm proximally, 1.79-2.45 mm at the midpoint, and 1.28-1.78 mm distally) on T1 and T3-DRIVE images than did other adult studies. Some authors suggested the following classification of PST: mild (3-3.9 mm), moderate (4-6.5 mm), or severe (> 6.5 mm) PST [5].
The actual incidence of pituitary stalk lesions in the general population remains unknown. Neoplastic diseases were reported as the most prevalent cause of pituitary stalk disorders, congenital and both inflammatory and infectious illness being noted to represent the second and third most common causes in children, respectively [6]. Congenital disorders and neoplasms remained prevalent in children under 10 years of age [7]. In children over 11 the frequency of infectious/inflammatory disorders was found to increase. A thickened pituitary stalk in patients with CDI necessitates a differential diagnosis that includes germinoma, Langerhans cell histiocytosis (LCH), lymphocytic hypophysitis (LYH) involving the infundibulum, neurosarcoidosis, tuberculosis, and adenoneurohypophysitis of another category, infiltration from adjacent neoplasms, and metastasis [8, 9]. Neoplastic infiltration and metastasis are radiographically distinguishable from regional and systemic findings. Chest X-ray studies and immunological and serological examinations provide diagnostic information for neurosarcoidosis and tuberculosis. Infundibulohypophysis may be distinguished from occult germinoma by the presence of an enhanced pituitary lesion and its frequent association with pregnancy in female patients [8]. However, LCH may take a course that is quite similar to occult germinoma.
Findings on the MRI of hypophyseal histiocytosis also demonstrated a homogeneously enhanced, thickened stalk and absent posterior pituitary hyperintensity [10]. Systemic bone lesions should be sought, but in cases of solitary hypophyseal lesions, obtaining a biopsy sample is mandatory. The cause of PST is usually identified within 3 years of surveillance. The definitive diagnosis is needed because these diseases are characterized by different methods of treatment and prognosis. The differential diagnosis between these diseases using tumour or biochemical markers and radiological examinations is difficult, and in most cases, only histopathological examination leads to the definitive diagnosis.
The reasons for PST in children and adolescents are presented below by frequency.
Intracranial germinoma
Epidemiology
According to the 2021 World Health Organization Classification of Tumours of the Central Nervous System (CSN) the types of germ cell tumour (GCTs) are: germinoma, embryonal carcinoma, yolk-sac tumour, choriocarcinoma, teratoma (mature, immature), teratoma with somatic-type malignancy, and mixed germ cell tumour [11]. Pure germinoma accounts for more than half of CNS GCTs in childhood. Patients with pure germinoma have a 10-year overall survival of over 90%. The median patient age is within the range of young adolescence, usually 10-14 years. A higher incidence is observed in Asian than in Western countries. CNS GCTs account for approximately 3–4% of primary paediatric brain tumours in the West, and 8–15% in Asia (based on data from Taiwan, Japan, and South Korea) [12]. The incidence rate in the United States is 0.1 per 100,000 person-years, and the incidence is 0.4-0.5 per 100,000 children per year in Taiwan [13, 14]. Three main germinoma locations may be distinguished. The most common site of origin is the pineal region (45%), and the second most common site is the suprasellar region (30%) within the pituitary stalk. A strong association between location and sex was noted in accordance with the literature, with the predominance of men in pineal lesions (75%) and of women in suprasellar lesions (70%) [15]. Approximately 5% to 10% of patients present with synchronous tumours arising in both the suprasellar and pineal locations, with the most frequent histology being a germinoma [16]. A rare phenomenon known as ectopic germinoma means that other areas of the CNS may be involved. They include the basal ganglia, ventricles, thalamus, cerebral hemispheres, and brain stem (5–10%) [17].
Signs and symptoms
The clinical presentation and time to diagnosis reflects the location and size of the tumour. Most patients with neurohypophyseal germinoma present endocrinological findings like polydipsia/polyuria, short stature, precocious puberty/delayed puberty, signs of central hypothyreosis, or central adrenal insufficiency. Additionally, the manifestations include ophthalmological ones (eye movement abnormalities, blurred vision, diplopia, visual field defect, ptosis, pupil dysfunction), neurological ones (headache, nausea/vomiting, dizziness/vertigo, and drowsiness), and motor impairment (dyskinesia/ataxia and focal/general weakness, facial palsy) [18]. According to Chang et al. [14], who described 49 children with intracranial pure germinoma in Taiwan, growth hormone (GH) deficiency or low insulin-like growth factor 1 (IGF-1) was diagnosed in 85.7%, adrenal insufficiency in 77.8%, CDI in 55.1%, central hypothyroidism in 48.4%, and hypogonadotropic hypogonadism in 44.4% of children.
CDI is the most common initial manifestation of neurohypophyseal germ cell tumour. Occasionally, CDI precedes the emergence of radiological abnormalities that indicate neurohypophyseal germinoma by several months or years. Such a germinoma-causing CDI was termed occult germinoma, which may result in a critical delay in the accurate diagnosis and, therefore, treatment of the disease [19]. Germinomas have an almost exclusive predominance as the occult cause of CDI in teenagers. Similarly to CDI, spontaneous and on stimulation, GH secretion deficiency often preceded the obvious tumour mass formation. Therefore, the association of CDI with evolving anterior pituitary endocrinopathy point to the presence of an occult neurohypophyseal germinoma. Czernichow et al. [20] conducted a study among 17 children with PST and CDI and diagnosed germinoma in 4 of them (15% of the patients). In another series of 9 children with PST and CDI, the transsphenoidal biopsy of PST showed germinoma in 7 of 9 patients (77.7%) [21].
Neurohypophyseal germinomas may be pure or contain syncytiotrophoblastic giant cells that secrete human chorionic gonadotropin (hCG). Any tumours affecting the neurohypophyseal or pineal region and producing hCG may cause pseudoprecocious puberty in boys [22]. Because hCG is structurally similar to luteinizing hormone (LH), its excessive increases may stimulate the Leydig cells of the testes to produce testosterone. Reduced follicle stimulating hormone (FSH) levels may explain the lack of correlation between testicular growth and other sexual characteristics, because testicular maturation depends upon this hormone. Precocious puberty in girls is extremely rare. Kitanaka et al. [23] described a 6-year-old girl who presented precocious puberty due to an hCG-secreting suprasellar immature teratoma. Starzyk et al. [24] presented a 5-year-old girl manifesting gonadotropin-releasing hormone-independent precocious puberty. The β-subunit human chorionic gonadotropin ( β-hCG) and α-fetoprotein (α-FP) tumour markers were markedly elevated, and suprasellar germ cell tumour was visualized with MRI [24]. It was believed that hCG did not trigger precocious puberty in girls in the absence of FSH, and it was used as an explanation for the rarity of precocious puberty in girls with hCG-secreting tumours. However, it was also reported that hCG was not only characterized by LH activity but also intrinsic, although weak, FSH-like activity [23]. It was also hypothesized that precocity was due to high aromatase activity in germ cell tumours [24].
Children with pineal lesions are at a high risk of presenting with acute hydrocephalus. In up to two-thirds of patients, lesions in the pineal region are associated with increased intracranial pressure and symptoms such as headache and vomiting, while Parinaud syndrome or upward gaze palsy are classic common manifestations seen in almost one-third of patients [18].
Germinoma arising from the basal ganglia, also known as ectopic germinoma, is a rare entity representing 5-10% of all intracranial germinomas. Basal ganglia are responsible for the control of movements, cognitive processes, emotions, and learning. Hemiparesis, cognitive decline, and behavioural disorders are common signs of basal ganglia germ cell tumour [25]. The ventricles, internal capsule, thalamus, putamen, cerebral hemispheres, and brain stem are other sporadic ectopic locations of GCTs.
The time from symptom onset to the diagnosis varies depending on the location of the tumour. The most prolonged interval was observed in case of suprasellar tumour (from one month to 9 years), the shortest in case of pineal tumour, and intermediate for a location in the basal ganglia (one month to 4.5 years) [26].
Diagnostics
GCTs may secrete measurable oncoproteins such as β-hCG and α-FP into the serum or cerebrospinal fluid (CSF). Although hCG is a glycoprotein with a molecular weight of about 45kD and has α- and β-subunits, germinoma may also independently secrete the β-subunits in the free form. β-hCG is produced by the normal trophoblast in the placenta. Serum levels are normally elevated in pregnant and postnatal women, and abnormally increased in patients with choriocarcinoma and germ cell, bladder, pancreatic, gastric, and lung tumours [27]. Tumour markers may be useful surrogate biomarkers and may spare some patients the need for biopsy. Typically, choriocarcinoma produces β-hCG, and yolk sac tumour produces α-FP. Germinoma with syncytiotrophoblastic giant cells also produces β-hCG, but its serum titre in germinoma with syncytiotrophoblastic giant cells is usually lower than in choriocarcinoma. Mildly elevated CSF β-hCG is detected in up to 38% of patients with suprasellar germinomas, whereas most of these patients have normal serum β-hCG concentrations. Mixed germ cell tumours produce various amounts of tumour markers according to the histological elements they contain. The markers may then be detected in both the serum and CSF. The assay of lumbar CSF is imperative for an accurate diagnosis and staging because oncoprotein levels are often much higher than in the serum [28].
The evaluation of the clinical diagnostic value of β-hCG and α-FP for GCTs is an interesting and confusing topic for endocrinologists, neurologists, and neurosurgeons.
The National Academy of Clinical Biochemistry emphasized that the detection of β-hCG should include the whole hCG and free β subunit at the same time. The recommendations of the International Society of Paediatric Oncology (SIOP) CNS GCT 96 protocol indicated that β-hCG concentration ≥ 50 IU/L and α-FP ≥ 25 ng/ml represented a strong indicator of the presence of germinoma. However, according to other authors, CSF concentrations that were much lower, but still greater than expected under normal conditions, were indicative of cerebral germinoma [29]. A cut-off value of β-hCG for the diagnosis of intracranial germinomas has not been established, thus limiting its diagnostic value. Institutions usually define their own normal range of serum β-hCG for intracranial germinomas. Commonly reported cut-off values range from 0.1 to 8 mIU/ml [30]. Zhang et al. [30] in their study including 93 intracranial germinoma and 289 intracranial non-germ cell tumour patients, recommended that the cut-off value of serum β-hCG of 0.5 mIU/ml could be used in assisting the diagnosis of germinoma, because it had the highest specificity (96.16%). Allen et al. [28] suggested the cut-off CSF β-hCG of 7.2 IU/l for germinoma and revealed that among 58 patients with histopathologically confirmed germinoma, approximately 34.5% of patients had elevated lumbar CSF β-hCG values with normal serum values, and in 87% of patients with hCG elevations abnormal CSF values were higher than the serum value. Hu et al. [31] proposed new biomarker diagnostic cut-off points: CSF β-hCG ≥ 8.2 IU/l and serum β-hCG ≥ 2.5 IU/l, while empirically adjusted criteria for α-FP are ≥ 3.8 ng/ml in the CSF and ≥ 25 ng/ml in the serum. According to the results published by Gonzales-Sanchez et al. [29] CSF β-hCG concentrations that exceeded the established reference interval (undetectable values to 0.7 IU/l) in the presence of suprasellar lesions and hypophyseal stalk thickening should be considered pathological, establishing the need to exclude the presence of germinoma. Katagami et al. [32] also claimed that a CSF/serum hCG ratio ≥ 2 represented a strong indicator of the presence of germinoma. However, it should be remembered that Hu et al. [31] reported a slightly increased level of β-hCG in the CSF in patients with LCH and with craniopharyngioma, with the respective values being 6 and 8 IU/l.
The determination of β-hCG in the serum or CSF is a sensitive indicator not only in the diagnosis of neurohypophyseal germinoma but also in monitoring treatment response and recurrence.
Other biomarkers associated with germinomas include lactate dehydrogenase and placental alkaline phosphatase. S-kit, a soluble product of the c-kit oncogene, appeared to be a more sensitive screening tool than hCG for germinoma in several series from Japan [33]. New markers were investigated, including CSF micro-RNA 371-373 and 302 levels, which were significantly increased in GCTs, but decreased significantly after treatment. This marker might be used for the future diagnosis of germinoma [34].
Despite the new diagnostic criteria proposed by the authors mentioned above, the surgical confirmation of tumour histology is still the standard of care in the absence of significant oncoprotein elevations ( β-hCG ≥ 50 IU/l, α-FP ≥ 25 ng/ml). A single exception should be mentioned, i.e. the presence of normal CSF and serum β-hCG and normal levels of α-FP supports the clinical impression of a germinoma in patients with bifocal germinoma, so histologic confirmation is not usually obtained.
Based on MRI findings, suprasellar lesions were classified into 5 types: a 3rd ventricle floor tumour extending dorsally with or without an identifiable pituitary stalk (Type Ia, Ib), ventrally (Type III), in both directions (Type II), a small lesion at the 3rd ventricle floor extending to the stalk (Type IV), and a tumour located in the stalk (Type V) [26]. The thickening of the hypophyseal stalk is the most common MRI finding, together with the loss of neurohypophyseal hyperintensity in T1-weighted sequences, in which the lesion appears isointense or slightly hyperintense with respect to the normal hypophysis. The uptake is less pronounced than in the normal hypophysis after gadolinium contrast injection [29]. The imaging findings are not specific, and differential diagnosis must encompass lymphocytic hypophysis, which is an infrequent disorder in children. Particular difficulty in the differential diagnosis is the fact that histological findings compatible with a lymphocytic inflammatory process may represent the first sign of a host reaction to occult germinoma [35]. This would justify the determination of hCG in the CSF in all prepubertal patients with a presumed or histological diagnosis of lymphocytic hypophysis, as well as the immunohistochemical study of the histological specimen with the determination of placental lactogen, c-Kit, and CD30 [36].
Secondary hypophysitis is believed to be triggered also by other focal pituitary processes, such as craniopharyngiomas, adenomas, and Rathke cleft cysts.
Treatment
The treatment includes chemotherapy (carboplatin, etoposide) and radiation therapy (18 Gy WV with a 12 Gy boost to the primary site). High-dose chemotherapy followed by autologous stem cell transplantation may be effective in patients with relapsed or progressive disease [37]. Because a number of patients are resistant or relapse after curative treatment, there is a pressing need for new therapeutic options. Genetic abnormalities that are present in such cases indicate that targeted therapies such as tyrosine kinase inhibitors may have promising activity. Results from a study evaluating dasatinib are awaited. CNS germinoma tumours may also express programmed death ligand-1 (PDL-1), which indicates the potential antitumour activity of immunotherapy. Additionally, the inhibition of the RAS/MAPK pathway may be a potential therapeutic option, but it requires further research [38]. Endocrine dysfunction manifested in germinoma cases might not be ameliorated even after undergoing successful therapy.
Surveillance and prognosis
Various authors emphasized that repeated brain MRI and the investigation of the tumour marker β-hCG in the serum and CSF should be performed every 3-6 months during the first 3 years after the onset of CDI and PST, in order to rapidly establish a diagnosis before the developing large tumour contributes to visual and neurological symptoms. Subsequently, MRI should be performed every year during the following 2 years and every 2–5 years thereafter, depending on the size and evolution of the lesion [20].
The prognosis of the tumour is dependent upon the histology, but also upon the size of the tumour and the extent of the disease at diagnosis. An early diagnosis is a key to treat such tumours before hypothalamic-hypophyseal damage proves irreversible or adjacent structures suffer compression or metastatic disease becomes apparent.
Cerebral histiocytosis
LCH is a rare disease of the monocyte-macrophage system characterized by the clonal proliferation of epidermal dendritic cells. In recent years, increasing evidence has been presented to support the idea that LCH is a “neoplasm”, which means that LCH tumours contain cells with gene mutations that cause them to make abnormal copies of themselves. A specific gene called the BRAF is mutated in about half of LCH tumours [39].
Epidemiology
LCH is a rare disorder, which mainly affects the paediatric population with peaks between 1 and 3 years of age. The estimated annual incidence of LCH is approximately 6:100,000 to 10:100,000 with the disease being significantly rarer in the population aged > 15 years (1:100,000) [40]. LCH is classified as localized or multifocal, and it may affect any organ or system of the human body. The clinical course may vary from a self-limiting disease to a rapidly progressive one that might lead to death. The disorder may involve the bones (78.7%), skin (36.7%), lung (25%), liver (25%), spleen (25%), lymph nodes (15%), and other soft tissues. The hypothalamus and pituitary gland are affected in 25% of patients [9].
According to the French Langerhans Cell Histiocytosis Study Group database, isolated CDI was the initial presentation of LCH in 26 patients, and the pituitary stalk was enlarged in 14 out of 1135 LCH patients under 18 years of age [41].
Signs and symptoms
Patients with LCH are often asymptomatic or present mild symptoms such as fatigue, generalized weakness, weight loss, night sweats, nausea, pruritus, and fever.
LCH in CNS exhibits a predilection for the hypothalamic-pituitary region (HPR), leading to permanent posterior and/or anterior pituitary hormone deficiencies in a subset of patients. HPR infiltration is present in 5-50% of children with LCH, but most commonly in those with the multifocal form [42]. An isolated presence of LCH is very rare in the HPR. The analysis of a large series of patients with LCH (1242 study subjects) revealed that only 0.04–0.6% had HPR involvement without extracranial lesions [43]. Isolated LCH of the infundibulum is rare, and such instances are typically reported as solitary cases in the literature [44]. CDI, the most frequent hormonal abnormality, occurs in 15–50% of patients with HPR LCH [42]. It may occur before, concurrently with, or many years after other multisystem manifestations of the disease that lead to the diagnosis of histiocytosis. Pituitary dysfunction occurs in only 5–20% of patients and is almost always accompanied by CDI (94%). The most common type of hormone deficiency in children and adults with HPR-involved LCH is GH deficiency (53–67%), followed by gonadotropin (53–58%), and thyroid-stimulating hormone (TSH) (3.9%) deficiencies. ACTH (adrenocorticotropic hormone) deficiency is observed in only 1–2%, while hyperprolactinaemia occurs in 40% of all HPR LCH patients [45].
Diagnostics
Currently, there is no specific biological marker of disease activity. As regards the differential diagnosis of PST, the following tests should be performed if LCH is taken into consideration: dermatological survey; laboratory evaluation including full blood count, erythrocyte sedimentation rate (ESR), alanine transaminase (ALAT), aspartate transaminase (ASPAT), gamma-glutamyl transpeptidase (GGTP), albumin, bilirubin level, international normalized ratio (INR), activated partial thromboplastin time (APTT); and radiographic evaluation including skeletal radiography survey, bone scintigraphy, chest radiograph, and ultrasonography of the abdomen to exclude other LCH lesions.
In a case of HPR LCH, an MRI scan usually shows an enlargement of the pituitary stalk with enhancement after gadolinium injection and the loss of spontaneous T1 hyperintensity of the posterior pituitary [41]. It is still difficult to differentiate HPR-involved LCH from other diseases, such as germinoma or lymphocytic hypophysitis with MRI. An isolated HPR lesion is a clinical challenge, because neither the clinical manifestations nor MRI may be sufficient to make a definitive diagnosis, and the risk associated with obtaining a diagnostic biopsy remains high. Nagasaki et al. [44] described a 13-year-old girl with CDI and PST and biopsy-confirmed LCH. The diagnostic work-up performed in this patient also revealed slightly elevated β-hCG in the cerebrospinal fluid, which indicated a difficulty not only with imaging but with biochemical differential diagnosis between neurosarcoidosis and germinoma.
Skeletal survey can be performed by multiple X-ray images and CT scans, but 18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG PET/CT) offers greater accuracy and allows a precise localization of lesions that are more easily accessible for biopsy than the pituitary stalk. 18F-FDG PET/CT is also a highly sensitive method for the staging and follow-up of paediatric patients with LCH.
The diagnosis of LCH should be based on the histological and immunophenotypic examination of the lesional biopsy. The presence of Birbeck granules under electron microscopy and positive immunohistochemical staining for S100 and CD1 and/or CD 207 (Langerin) protein markers is characteristic [46].
Treatment
Very low incidence of localized HPR LCH is the main reason why there is no consensus as to optimal treatment. The treatment of isolated HPR LCH is controversial and individualized. Simple observation, surgery, low-dose radiation, and chemotherapy (glucocorticoids, methotrexate, vinblastine) are considered in treatment planning. Limited literature data show that low-dose irradiation (≤ 22 Gy) is usually the first-line therapy adequate for most cases of isolated HPR LCH with a sporadic recurrence of the illness [47].
The natural history of LCH is poorly understood. In fact, spontaneous regression is well reported in numerous body systems including the skin, bone, lung, and CNS [44].
Lymphocytic hypophysitis
LYH is a neuroendocrine disorder characterized by autoimmune inflammation of the pituitary gland with various degrees of pituitary dysfunction. It was initially considered to be restricted to women in relation to pregnancy, but it is now clear that LYH may occur in children and adults, men and women [48].
The following types of LYH may be distinguished: lymphocytic adenohypophysis (LAH – an inflammation limited to the anterior hypophysis), lymphocytic infundibuloneurohypophysitis (LINH – autoimmune infiltration involving the infundibular stem and posterior lobe), and lymphocytic panhypophysitis (LPH – both the adenohypophysis and the infundibuloneurohypophysis are affected), depending on which part of the HPR is involved in the immune process. LAH was first described in 1962 by Goudie and Pinkerton [49]. They reported a case of a 22-year-old woman, who died 14 months after her second delivery, probably because of adrenal insufficiency. LINH was first described in 1970 by Saito et al. [50], who observed a 66-year-old asthmatic woman with a one-month history of polydipsia and polyuria. LINH was reported especially in children and adolescents. LPH was first described in 1991 in a 40-year-old man with a 3-month history of headache, polydipsia, polyuria, and impotence [51].
Epidemiology
The annual incidence of LYH may be estimated at one case per 9 million. Data from 4 large patient analyses (905–2500 people) indicate that LYH is seen in less than 1% (0.88-0.24%) of all surgical pituitary specimens [52–54]. In 2012, Kalra et al. [55] indicated that the number of children under 18 years old with LYH was greater than 96 in the literature. The incidence in children was much lower compared to approximately 460 adults with autoimmune hypophysitis [56]. In contrast to adults (more female patients were identified with a female-to-male ratio of 8:1) no significant sex predilection was seen among children.
Concurrent autoimmune conditions were reported in 20–50% of cases of LYH in adults [57]. The most common association was observed with autoimmune thyroid disease, reported in 15–25% of LYH cases, i.e. 70-80% of cases with an associated autoimmune disease. Chronic autoimmune thyroiditis constituted about 75%, while Graves’ disease and subacute thyroiditis were reported less frequently. Autoimmune adrenalitis was reported in 5–7% of cases (15–25% of patients with an associated autoimmune condition), while pernicious anaemia and type 1 diabetes were seen in 2% of cases [57]. Associated autoimmune disorders occurred in 7 out of 96 children, with an incidence of 7% as compared to 20–50% in adults [55].
Signs and symptoms
The clinical presentation of LYH is variable and comprises 4 categories of symptoms: sellar compression, hypopituitarism, CDI, and hyperprolactinaemia. In adults with LYH, 96% have PST and 72% have CDI.
The symptoms of sellar compression, represented by headache and visual disturbances, are the most common and usually the initial complaints in LAH. Headache is the result of the distortion of the dura mater and diaphragma sellae by the expanding pituitary mass. However, a recent study showed that pituitary volume did not correlate with its severity [58]. Headache is observed more often in patients with LAH (53%) than in those with LINH (13%). Visual abnormalities include visual field defects, decreased acuity, and diplopia, which are secondary to the compression of the optic chiasm by a pituitary mass expanding upward or laterally to the cavernous sinus. Visual disturbances are described in 43% of LAH and seldom in LINH, i.e. only in 3%. The other most common symptoms are due to a partial or complete deficit of the anterior pituitary hormones. CDI is another of the most common problems, which may be attributed either to direct immune destruction or to the compression of the posterior lobe and infundibular stem. CDI is the cardinal feature of LINH (98%) and may be very rare in LAH (1%). CDI may be masked in the presence of a coexisting glucocorticoid deficit because it opposes the action of antidiuretic hormone (ADH) at several levels. Glucocorticoids inhibit the secretion of ADH from the neurons of the paraventricular nucleus and suppress the synthesis of aquaporin 2, an ADH-dependent water channel expressed in the renal collecting tubules. The manifestations of hyperprolactinaemia, mainly represented by amenorrhoea and galactorrhoea, are the least common. Hyperprolactinaemia results from stalk compression, with the resulting decrease in dopamine delivery to the anterior pituitary. The inflammatory process may directly destroy the lactotrophs, inducing the release of prolactin (PRL) into the general circulation [59]. Headache, visual disturbances, hypocortisolism, and hypothyroidism are more common in LAH than in LINH or LPH. In contrast, CDI suggests LINH, a condition in which other presenting symptoms, such as visual disturbances, hypocortisolism, hypogonadism, and hyperprolactinaemia are rare.
Diagnostics
A presumptive diagnosis of LYH may be made based on clinical, laboratory, and imaging studies, but the definitive confirmation requires a pituitary biopsy.
Antibodies against pituitary protein and vasopressin-secreting hypothalamic cells may be found in patients with LYH. They may be measured with indirect immunofluorescence, immunoblotting, or enzyme-linked immunosorbent assay (ELISA). However, the specificity and sensitivity of pituitary antibodies is poor, as they were found in various pituitary diseases such as Cushing’s disease, pituitary adenoma, empty sella syndrome, as well as in other autoimmune diseases, such as type 1 diabetes mellitus, Hashimoto’s thyroiditis, and Graves’ disease. A retrospective analysis of 379 patients with LYH conducted by Caturegli et al. [51] revealed that antibodies against pituitary protein were detected in 10–80% of patients depending on the form of LYH and the method used (they were more often confirmed in immunoblotting).
The authors of a case series presenting LAH reported that MRI studies showed an enlarging pituitary mass in 75-90% of patients [48]. Even in cases with CDI, in which the initial scan was normal, repeated imaging evaluations performed months later might reveal a mass-like image [60]. Heinze and Bercu [61] published a review of 63 patients in which they reported a post-contrast enhancement of the lesion in 70%. Honegger et al. [54] reported a tongue-like extension toward the hypothalamus as a characteristic finding of LAH. A diffuse thickening of the pituitary stalk is a very characteristic MRI feature in patients with LINH. Its greater diameter was found to exceed 3.5 mm at the level of the median eminence of the hypothalamus [62]. The normal smooth tapering of the infundibular stalk is lost, and a varying degree of asymmetry may exist. Marked gadolinium enhancement of the stalk is quite common, extending even into the lower hypothalamus. The loss of the usual neurohypophyseal “bright spot” was also commonly reported. However, it should be kept in mind that this MRI sign may be absent in 10% of normal subjects [63]. Extensive pituitary inflammatory changes were reported in LINH, extending upwards to the suprasellar area and affecting the optic pathway, or laterally to the cavernous sinuses, with thickening of the pituitary stalk, enlargement of the neurohypophysis, or lack of the neurohypophyseal bright spot on the normal neurohypophysis on MRI [64]. Histopathology remains the gold standard for the diagnosis of LYH. The infiltration of the pituitary gland with lymphocytes, with the predominance of T cells and particular CD4 cells is the defining pathological feature of LYH. Plasma cells, eosinophils, macrophages, histiocytes, and neutrophils are also present. Fibrosis and rare necrosis may be reported in pathological specimens [63].
LINH should be suspected as a cause of PST when we observe: an acute onset of CDI with headache and mass-effect symptoms; isolated, early or disproportionate disruption of ACTH or TSH secretion, disproportionate disruption of anterior pituitary function for the magnitude of the changes on MRI; the presence of other autoimmune conditions and/or positive autoantibodies, including thyroid peroxidase antibodies, antinuclear antibodies, antiparietal cells, adrenal antibodies, and anti-smooth muscle antibodies; the presence of antipituitary antibodies in the serum; lymphocytic pleocytosis in the CSF; and characteristic MRI findings (diffuse thickening of the pituitary stalk with or without enhancement after gadolinium, loss of the normal posterior “bright spot” on T1-weighted images) [51].
Treatment
Currently, the treatment of LYH is only symptomatic. It includes reducing the size of the pituitary/stalk mass and/or replacing the defective endocrine function. Mass reduction may be achieved via pituitary surgery, immunosuppressive drugs, or radiotherapy. Glucocorticoids should be used as the first line of treatment, rather than surgery. Glucocorticoids may be effective for treating LYH, both as anti-inflammatory agents to reduce the size of the pituitary mass or the thickened stalk, and as the replacement of defective adrenal function. Prednisone, hydrocortisone, and methylprednisolone were the most commonly used glucocorticoids [65]. In the case of patients with poor response to glucocorticoids, other immunosuppressive drugs, such as azathioprine, methotrexate, or cyclosporine A, may be used [53]. Surgery should be performed only in the presence of serious and progressive deficits of visual field, visual acuity, ocular movements, or increased intracranial pressure, not responsive to pharmacological treatment. Radiotherapy (conventional fractionated external-beam radiotherapy, Gamma Knife radiosurgery) should be limited to cases with severe mass-effect symptoms, those who show poor response, or those who are poor candidates for high doses of glucocorticosteroids and/or surgery. However, it is important to mention the findings of some authors, e.g. Caturegli et al. [51], who reported that LYH resolved spontaneously without any treatment in 11 patients (3%).
Neurosarcoidosis
Sarcoidosis is an auto-inflammatory, granulomatous disorder of unknown aetiology, with multi-organ involvement, hilar lymphadenopathy, and pulmonary interstitial infiltrations being the most common manifestations. Furthermore, it affects the eyes, skin, liver, and spleen. Sarcoidosis may also involve any area of the CNS: the meninges, ventricles, cerebellar hemisphere, white matter of the frontal lobe, adjacent brain or spinal cord parenchyma, pituitary stalk, pituitary, hypothalamus, thalamus, optic nerve and chiasm, cranial nerves such as the facial, auditory, and vestibulocochlear nerve.
Epidemiology
The incidence of clinically recognized sarcoidosis in children is 0.22–0.29/100,000 children per year, and gradually increases with age to a small peak in teenagers at 13–15 years of age [66]. However, adults aged 45–55 years are most often affected, with half of them being older than 55 years of age at the time of diagnosis. Neurosarcoidosis was reported in 5–10% of adults with systemic sarcoidosis and was seldom recognized in children [67]. Isolated neurosarcoidosis is very rarely diagnosed. According to the English sources, a total of 54 children and adolescents were diagnosed with neurosarcoidosis, and only 9 children with isolated neurosarcoidosis. The average age of onset was 12 years (3 months – 18 years) [68].
Signs and symptoms
General symptoms of sarcoidosis are fatigue, weight loss, fever, persistent cough, skin changes (subcutaneous infiltrates, erythema nodosum, lupus pernio), eye lesions (uveitis, retinal changes, conjunctival nodules), and peripheral lymphadenopathy.
The manifestations of neurosarcoidosis include seizures, headache, vomiting, somnolence, cranial neuropathies (common: VII, II, infrequent: III, IV, VI, V, VIII), pituitary and hypothalamic dysfunction, cerebellar signs, neuropsychological deficits, myelopathy, and peripheral neuropathy. Granulomatous masses that involve the hypothalamus and pituitary gland cause the following: diabetes insipidus, growth and sexual maturation failure, and syndrome of inappropriate antidiuretic hormone secretion (SIADH). Hypothalamic-pituitary involvement accounts for 6–9% of all neurosarcoidosis cases. Endocrine investigations mostly identify GH, gonadotropin and thyrotropin deficiency, CDI in over half of cases, and – the least frequently – corticoadrenal insufficiency [69]. Convulsions are the most common symptom of neurosarcoidosis in children, while cranial neuropathies, especially in nerve VII, are the most common after puberty and in adults [68].
Diagnostics
Neurosarcoidosis is a diagnosis of exclusion. There is no single diagnostic test for sarcoidosis. Diagnosis is achieved by the combination of history, examination, CNS MRI, CSF analysis, gallium-67 scintigraphy, 18F-FDG PET/CT, and biopsy. The MRI features of neurosarcoidosis include diffuse leptomeningeal thickening and enhancement, focal dural or brain parenchymal enhancement with or without mass effect, periventricular radial vascular enhancement, and enhancement, enlargement, or atrophy of the cranial nerves or enlargement of the pituitary stalk. Criteria for the paediatric diagnosis of neurosarcoidosis have not been established, although most clinicians use the classification scheme (definitive-probable-possible) by Zajicek et al. [70]. Anaemia, leukopaenia, and eosinophilia are commonly seen on blood counts [71]. Hypercalcaemia and/or hypercalciuria occur in up to 10% and 40% of children, respectively, because a sarcoid macrophage is able to synthesize 1,25-dihydroxyvitamin D [72]. Angiotensin-converting enzyme (ACE) is a biomarker of sarcoidosis. ACE levels are elevated in sarcoidosis because of the activation of monocytes, which are the precursors to the epithelioid cells that form granulomas. ACE is not specific for sarcoidosis. However, it is elevated in the serum of 68-88% of patients with systemic disease and in the CSF of 24–55% of patients with neurosarcoidosis [73]. It should be noted that other disease processes that have also shown elevated serum ACE levels include tuberculosis, lung cancer, Hodgkin lymphoma, and cirrhosis of the liver. Normal serum/CSF ACE level cannot exclude the diagnosis of sarcoidosis. However, an elevated CSF ACE level should prompt further neurosarcoidosis evaluation.
The analysis of the CSF may show pleocytosis (> 5 white blood cells) with slight lymphocytosis (10–100 cells per µl), slightly elevated protein (> 50 mg/dl) in 76% of samples, slightly decreased glucose (< 50 mg/dl) in 51% of samples and increased immunoglobulins with oligoclonal banding [73]. However, CSF abnormalities are not specific to neurosarcoidosis, and normal CSF results were obtained in more than a third of cases [70]. A retrospective review of the Mayo Clinic record system revealed that oligoclonal banding was present in the spinal fluid of 18% of patients [74].
Kidd et al. [72] presented 6 patients with hypothalamic involvement of neurosarcoidosis in MR imaging. Stalk enlargement was reported in 2 of those patients.
In a case of HPR involvement, a whole-body gallium or FDG-PET scan can show other lesions that are more accessible for biopsy.
Biopsy still provides the strongest evidence for this disease. The diagnosis of sarcoidosis is confirmed by demonstrating a typical noncaseating granuloma on a biopsy specimen.
Treatment
No established treatment guidelines exist for neurosarcoidosis. High-dose corticosteroids, particularly oral prednisone, are widely used and are often effective in the treatment of sarcoidosis. If corticosteroid therapy is unsuccessful, second-line immunosuppressants are administered, e.g. methotrexate, azathioprine, mycophenolate mofetil, and leflunomide [75]. More recently, third-line agents, including tumour necrosis factor inhibitors, i.e. infliximab or adalimumab, have been indicated in the treatment [75]. Infliximab was dentified as an effective treatment option for patients, including those who had previously been refractory to other immunosuppressants [71].
Pituitary tuberculosis
Epidemiology
Pituitary tuberculosis should also be considered in the differential diagnosis of PST. The incidence of tubercular hypophysitis is 0.5–4% of all intracranial lesions. 25–30% of tubercular hypophysitis cases reported in the literature had previous or active tuberculosis. Primary pituitary tuberculosis caused by Mycobacterium tuberculosis is a sporadic condition, and only 106 cases were reported from 1924 to 2019 [76]. It mainly affects young adults with the median age being 35 years (from 5 to 60). Several cases of children with pituitary tuberculosis were also reported, especially in endemic countries.
Signs and symptoms
Headache (85.2%), somnolence, visual disturbances (48.1%), low-grade fever (14.8%), and vomiting are common clinical symptoms. Anterior pituitary insufficiency (51.9%), hyperprolactinaemia (27.6%), and CDI are common endocrine dysfunctions in pituitary tuberculosis [77]. The presence of CDI is one of the key features in distinguishing pituitary tuberculoma from pituitary adenoma.
Diagnostics
It is difficult to differentiate inflammatory lesions from those of pituitary adenoma clinically and radiologically. In MRI, pituitary tuberculosis usually presents the features suggestive of a sellar and suprasellar mass, and it may be very difficult to differentiate between pituitary tuberculoma and adenoma [78]. The thickening and nodularity of the pituitary stalk are also considered to be signs of pituitary tuberculoma. However, this finding is non-specific; it is also seen in other inflammatory conditions like sarcoidosis or lymphocytic hypophysitis.
The following tests should be performed in patients with a suspected tuberculous aetiology of PST: tuberculin skin test, QuantiFERON-TB test (sensitivity 92.6%), and chest X-ray/computed tomography (CT). CSF examination may reveal moderate pleocytosis (100–300 mm3), increased protein level (100–400 mg/dl), and decreased glucose level (< 40 mg/dl). The samples do not always stain for acid-fast bacilli and Mycobacterium spp. when tested with polymerase chain reaction (PCR) method [76]. The definitive confirmation of the diagnosis requires a histopathological examination, which reveals necrotizing granulomatous inflammation, staining for acid-fast bacilli (not usually demonstrable from pathological tissue), and the PCR of Mycobacterium spp. in DNA extracted from the tissue.
Treatment
Surgery is not indicated in tuberculous hypophysitis except for obtaining biopsies to confirm the diagnosis. Antitubercular drugs that cross the blood-brain barrier are administered to patients for 9–24 months depending on the clinical and imaging outcome [77]. A significant reduction in size was reported as early as 2 months after treatment, usually with a complete resolution of sellar mass at the end of the regimen. The improvement of endocrinological status and even eliminated panhypopituitarism were reported in some patients [79]. In cases such as tubercular hypophysitis, early diagnosis is very important given that panhypopituitarism may be eliminated in clinical terms following an effective anti-tuberculosis treatment. A delayed diagnosis may lead to permanent endocrine dysfunction.
Diagnostic evaluation of patients with PST
Considering the causes of PST described above, Table 1 presents tests used in the diagnostic process of PST in children.
After PST imaging in MRI, diagnostic tests should be planned, taking into account the history and physical examination data. First- and second-line tests may be distinguished.
Table I
Diagnostic test of pituitary stalk thickening in children
[i] PST – pituitary stalk thickening; TSH – thyroid–stimulating hormone; FT4 – free thyroxine; MRI – magnetic resonance imaging; hCG – human chorionic gonadotropin; α-FP – α-fetoprotein; LCH – Langerhans cell histiocytosis; ACTH – adrenocorticotropic hormone; PLAP – placental alkaline phosphatase; CSF – cerebrospinal fluid; aTPO – thyroid peroxidase antibodies; LH – luteinizing hormone; FSH – follicle–stimulating hormone; E2 – oestradiol; T – testosterone; LH-RH test – luteinizing hormone–releasing stimulation test; ANCA – anti-neutrophil cytoplasmic antibodies; ALAT – alanine transaminase; ASPAT – aspartate transaminase; GGTP – gamma–glutamyl transpeptidase; LDH – lactate dehydrogenase; PRL – prolactin; INR – international normalized ratio; APTT – activated partial thromboplastin time; IGF-1 – insulin-like growth factor 1; IGF–BP3 – insulin-like growth factor–binding protein 3; GH – growth hormone; Ca – calcium; 1,25OHD – 1,25-dihydroxyvitamin D; PET – positron emission tomography; CT – computed tomography; 18F–FDG PET/CT – fluorodeoxyglucose positron emission tomography/computed tomography; ACE – angiotensin–converting enzyme
The first-line tests used in the diagnosis of patients with PST include: complete blood count, ESR, ALAT, ASPAT, GGTP, urea level, creatinine, INR, APTT, blood electrolytes; 1,25-dihydroxyvitamin D; serum ACE, QuantiFERON, tuberculin test; antipituitary antibodies, anti-vasopressin antibodies, systemic autoantibodies; endocrine tests assessing the function of the anterior and posterior pituitary gland, serum tumour markers ( β-hCG, α-FP); imaging tests (chest X-ray, skeletal radiograph, abdominal ultrasound), and neurological and ophthalmic examination.
If first-line tests are inconclusive, it is possible to use the so-called second-line tests in selected cases, depending on the clinical presentation. These tests include the following: tumour markers in CSF, ACE in CSF, the detection of Mycobacterium spp. by PCR in CSF, and immunoglobulin G4 (IgG4) in the serum. Imaging tests of the second line are whole-body imaging with MRI, gallium 67-scintigraphy, 18F-FDG PET/MRI, or PET/CT. It is crucial when LCH or sarcoidosis is suspected because it might detect extracranial lesions not otherwise identified, which may be biopsied.
In 2021, the British national recommendations for diagnosing children and adolescents with PST, CDI, or both, developed by specialists under the auspices of paediatric endocrinology and oncology societies, were published [80]. According to the recommendation mentioned above, a routine CSF examination for acid-fast bacilli is not recommended due to the sporadic occurrence of pituitary tuberculosis in children. Nonetheless, it should be considered in exposed patients or those at a high risk. Likewise, due to the sporadic diagnosis of neurosarcoidosis as the cause of PST in children and adolescents, the authors do not recommend routine ACE in the CSF, unless neurosarcoidosis is strongly suspected. Because IgG4-related hypophysitis seldom affects a child or young person, routine measurement of IgG4 concentrations is not currently advisable.
The indications for and timing of endoscopic pituitary biopsy in children and young people are controversial. Biopsy should be performed in a specialist pituitary neurosurgical unit with perioperative endocrine support on-site to reduce the risk of complications. According to the British recommendations of 2021, pituitary biopsy should be considered only in markedly thickened (≥ 6.5–7 mm) or enlarging stalk with additional visual or endocrine deficits, which continue to pose a worrying diagnostic dilemma after repeated tests [80]. Histopathological examination is conclusive in most cases. However, it is important to remember that histological findings compatible with a lymphocytic inflammatory process may represent the first sign of a host reaction to occult germinoma, which requires a repeated biopsy [35]. In the case of CDI, a repeated brain MRI and investigation of β-hCG tumour marker in the serum and CSF should be performed every 3–6 months during the first 3 years after the onset of CDI (the frequency depending on the presence of a progressively enlarging pituitary stalk), to rapidly establish a diagnosis before the developed large tumour triggers visual and neurological symptoms. Subsequently, MRI should be performed every year for the following 2 years and every 2–5 years thereafter, depending on the size and evolution of the lesion [20]. Therefore, the follow-up strategy should be individually adapted.
Conclusions
The spectrum of pathology involving the pituitary infundibulum is broad. The pituitary infundibulum presents a diagnostic imaging challenge because of its small size and the protean spectrum of disease processes.
CNS tumours, especially germinoma, are the most common causes of PST in children. Germinoma should be suspected in all children with PST specially with CDI or pituitary deficiency, even when neurological and ophthalmological symptoms are absent. Young age, male gender, a very large thickening of the stalk, a higher number of anterior pituitary deficits, as well as a serum level of prolactin above 1.3 times the upper limit of normal are all non-invasive parameters increasing the likelihood of a neoplastic origin [81]. Histiocytosis or LYH are rarely diagnosed. Neurosarcoidosis, granulomatosis, or specific inflammations are only described in the paediatric population as case studies. Despite the progress in biochemical and radiological diagnostic testing, the final diagnosis of a stalk lesion or sellar tumour is still based on a histopathological examination. In the case of sarcoidosis or histiocytosis, the histopathological examination may concern peripheral tissues (if it is not an isolated form).
Patients should be diagnosed and managed in specialist paediatric endocrine centres with experience in pituitary diseases. The cooperation of many specialists, i.e. endocrinologist, radiologist, oncologist, and neurosurgeon, is necessary.









