
Introduction
In the past few decades, mortality caused by gastric cancer has decreased [1], but gastric cancer is still the second leading cause of cancer mortality in the world [2]. Because metastasis is the most important cause of death, many efforts have been focused on better understanding of its mechanisms. Emerging evidence suggests that epithelial-mesenchymal transition (EMT) is quite important in the metastasis of various kinds of solid tumours [3, 4], including gastric carcinomas [5]. However, the mechanism that leads tumours to induce EMT is poorly understood.
Epithelial-mesenchymal transition is a coordinated molecular and cellular change defined as a reduction in cell-cell adhesion, apical-basolateral polarity, and epithelial markers, as well as an acquisition of motility, spindle-cell shape, and mesenchymal markers [6]. This process involves a disassembly of cell-cell junctions, including downregulation and relocation of E-cadherin as well as upregulation and relocation of N-cadherin [7]; hence, both E-cadherin and N-cadherin are important molecular markers of EMT. Also, the processes of EMT can be triggered by various growth factors, such as platelet-derived growth factor-B (PDGF-B) [8–10]. Recently, Michiyo Kodama et al. [11] showed that PDGF-B secreted by tumour cells and PDGFR-b expressed by stromal cells are associated with lymphatic metastasis in gastric carcinoma, and they inferred that the tumour microenvironment might play quite an important role in PDGF-B-induced tumour metastasis. However, the mechanism by which PDGF-B signalling promotes metastasis in human gastric carcinoma is still unknown. Also, MAPK/ERK signalling, an active tumour metastasis-relating pathway, is widely reported to be activated by many kinds of growth factors to promote EMT of tumour cells [12, 13]. So, we conjectured that PDGF-B signalling might induce EMT to promote metastasis via activation of MAPK/ERK signalling in human gastric carcinoma.
In our study, we established two gastric carcinoma cell lines, MKN28 and MKN45, to stably overexpress PDGF-B by lentiviral vectors, and detected their expressions of E-cadherin, N-cadherin, and ERK-1. Then, PDGF-B overexpression MKN28 and MKN45 cells were cocultured with PDGFR-b-positive fibroblast (hs738) cells to construct the activation of PDGF-B signalling. After that the MAPK inhibitor was added. Finally, we detected their expressions of E-cadherin, N-cadherin, and ERK-1 to ascertain whether PDGF-B signalling promoted EMT and its mechanism.
Material and methods
MAPK inhibitor
MAPK inhibitor VIII, Isozyme-Selective, ERKi-1/2 (Santa Cruz Biotechnology Inc., USA) was used in this study at a concentration of 0.1 µg/ml.
Cell culture
MKN28 and MKN45 human gastric carcinoma cell lines and fibroblast (hs738) were provided by the Cell Bank of the Chinese Academy of Sciences, Shanghai, China. Cells were cultured in Roswell Park Memorial Institute 1640 (RPMI1640) media supplemented with 10% foetal bovine serum (FBS).
Lentiviral vector constructs and preparation
A lentiviral-delivered PDGF-B vector was constructed and prepared by Chongqing Western Technology Inc. (Chongqing, China) as described by Lois et al. [14] and Xia et al. [15]. Briefly, primers were designed according to the PDGF-B sequence (Genbank Accession Number NM_002608.2). The following primer sequences were used: PDGF-B-F, 5’- ATGAATCGCTGCTGGGCGCTC-3’; PDGF-B-R, 5’-CTAGGCTCCAAGGGTCTCCTTC-3’. The target gene was obtained by polymerase chain reaction (PCR) and was inserted into the pUC57 vector. Then, both the pLenO-DCE and pUC57-PDGF-B were enzyme digested by EcoR I and Not I, respectively. After ligation, the pLenO-DCE-PDGF-vector was constructed. After sequencing, the pLenODCE-PDGF-B vector was transfected into 293T cells and lentiviral-delivered PDGF-B vector was prepared.
Cell transfection
Briefly, 1 × 105 MKN28 and MKN45 cells were seeded in each well of a 6-well plate in 500 µl of complete media at 37°C in a 5% CO2 incubator for 24 h, and then transduced by lentiviral vectors at a multiplicity of infection of 10 : 1 [16]. Transduction was carried out in the presence of Polybrene (8 µg/ml). After washing 3 times with PBS, 1 ml of RPMI1640 was added to each well. Cells were seeded at 37°C in a 5% CO2 incubator for 48 h. Fluorescence microscopy was used to observe the transduction. 400 µg/ml of green fluorescent proteins was used for screening. Transduced cells were passaged and seeded for further experiments.
Cell coculture
Stratified co-culture technique was used for cell co- culture [17]. Normal gastric carcinoma cells (MKN28 and MKN45) and fibroblast (hs738) cells, PDGF-B overexpression gastric carcinoma cells (MKN28 and MKN45), and fibroblast (hs738) cells were cocultured (1.5 × 105 of each cell type) in 6-well Transwells (Corning Inc., Corning, NY) for 72 h [17]. The fibroblast (hs738) cells were plated in RPMI1640 (10% FBS) at 37°C on the underside of the Transwells. After 90 min, the Transwells were reinserted into the 6-well plate and normal gastric carcinoma cells (MKN28 and MKN45) or PDGF-B overexpression gastric carcinoma cells (MKN28 and MKN45) were plated in the upper chamber of the Transwell. After being cocultured for 72 h,normal gastric carcinoma cells (MKN28 and MKN45) or PDGF-B overexpression gastric carcinoma cells (MKN28 and MKN45) were gathered.
Western blot analysis
Cells were lysed on ice in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 2 mM sodium fluoride, 2 mM Na3VO42, 1 mM EDTA, and 1 mM EGTA). Total protein extracts were analysed by western blotting, as described previously [18]. Proteins (20 µg) were separated by SDS-PAGE gels (Invitrogen) and transferred to PVDF membranes. The membranes were blotted for 1 h with 5% milk. Membranes were incubated with primary antibodies (1 : 500 dilution) against E-cadherin, N-cadherin, or ERK-1 (Santa Cruz Biotechnology, Inc., USA) at 4°C overnight. After incubation with horseradish peroxidase-conjugated secondary antibody (1 : 1000 dilution) for 3 h at 37°C, signals were detected by ECL chemiluminescence for 5 min. The films were analysed by densitometry with image software.
Results
Construction and evaluation of platelet-derived growth factor-B overexpression MKN28 and MKN45 gastric carcinoma cells
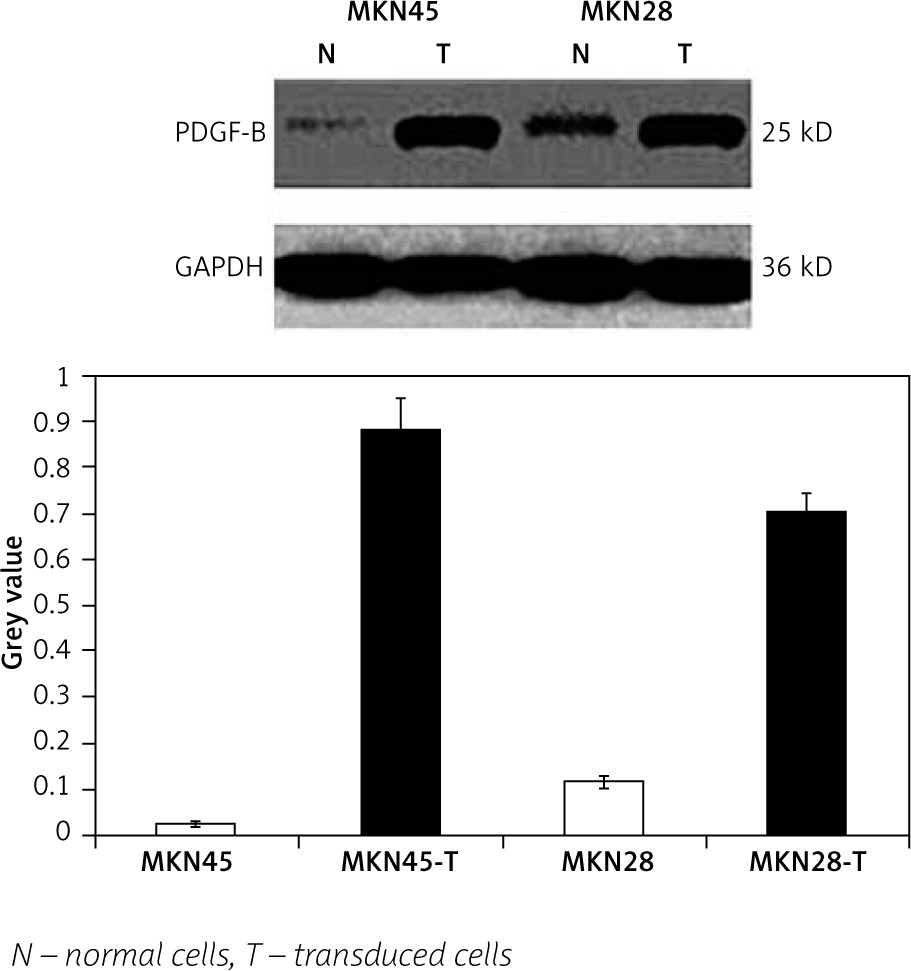
To check the overexpression of PDGF-B in our stably transfected cells, we checked the immunofluorescence and expression of PDGF-B protein in our transfected MKN28 and MKN45 gastric carcinoma cells, which were transduced with lentiviral constructs to overexpress PDGF-B. As illustrated in Fig. 1, our stably transduced cells showed significant green fluorescence. Also, the protein levels of PDGF-B increased significantly in transfected cells (p < 0.05) (Fig. 2).
Fig. 1
Immunofluorescence of MKN45 and MKN28 gastric carcinoma cells after transfection of platelet-derived growth factor-B lentiviral vector (× 200)
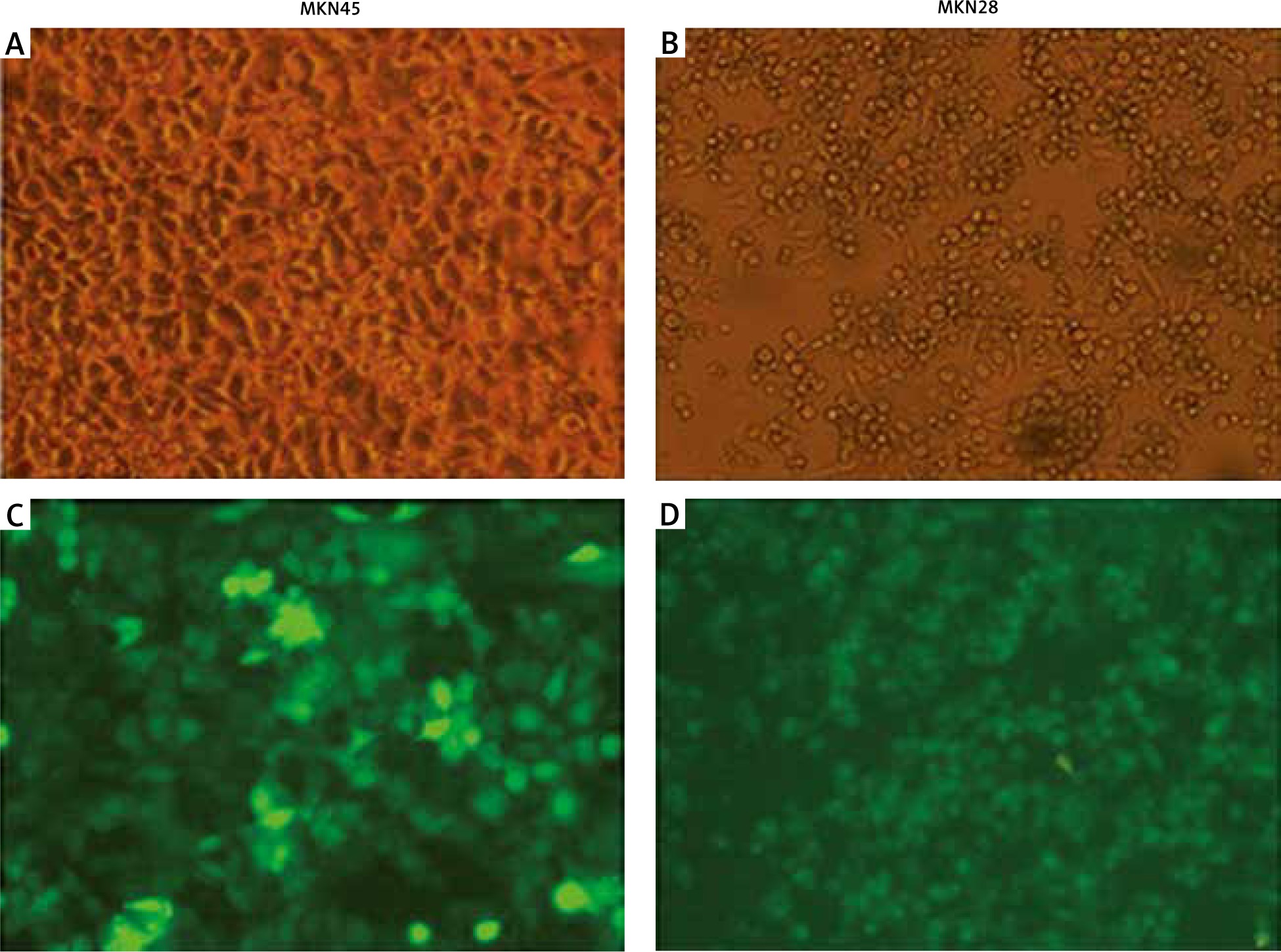
Fig. 2
Platelet-derived growth factor-B protein high expression in transfected MKN45 and MKN28 gastric carcinoma cells
Expression of E-cadherin, N-cadherin, and ERK-1 in platelet-derived growth factor-B overexpression and normal MKN28 and MKN45 gastric carcinoma cells
We determined the expressions of E-cadherin, N-cadherin, and ERK-1 protein in PDGF-B overexpression MKN28 and MKN45 cells and normal MKN28 and MKN45 cells by western blot analysis. As illustrated in Fig. 3, expressions of E-cadherin, N-cadherin, and ERK-1 protein were no different between PDGF-B overexpression MKN28 and MKN45 cells and normal MKN28 and MKN45 and cells (p > 0.05) (Fig. 3).
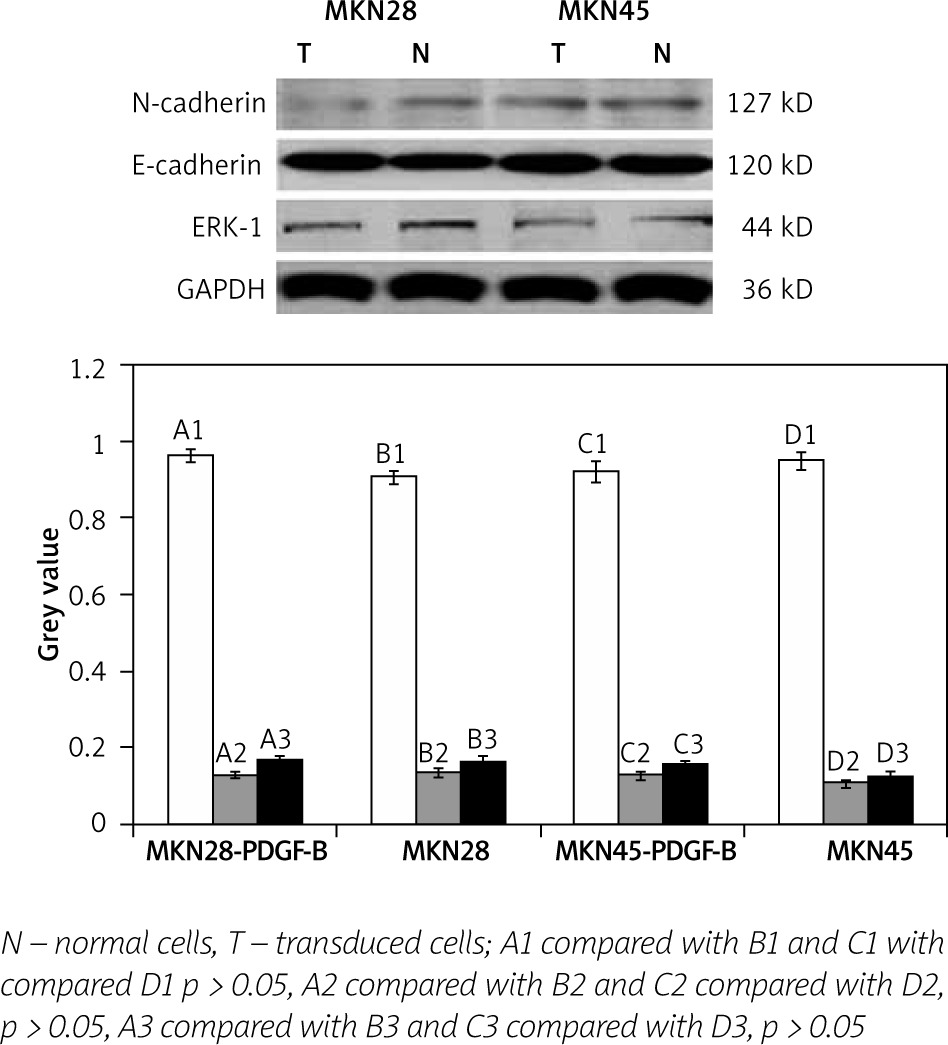
Expression of E-cadherin, N-cadherin, and ERK-1 in platelet-derived growth factor-B overexpression and normal MKN28 and MKN45 gastric carcinoma cells after coculture
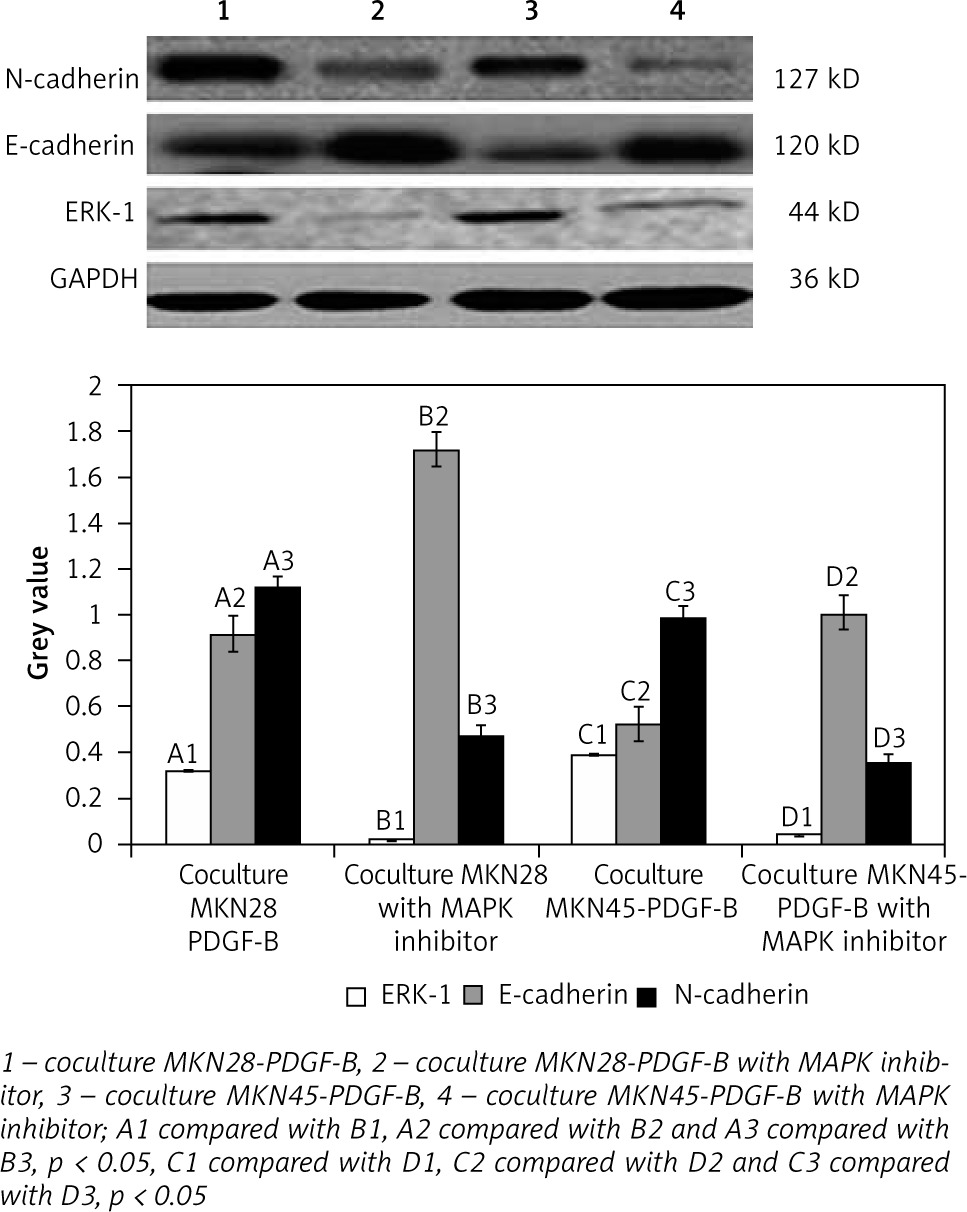
For activation of PDGF-B signalling, both normal gastric carcinoma cells (MKN28 and MKN45) and PDGF-B overexpression gastric carcinoma cells (MKN28 and MKN45) were cocultured with fibroblast (hs738) cells. Then, expressions of E-cadherin, N-cadherin, and ERK-1 protein in PDGF-B overexpression and normal MKN28 and MKN45 cells were detected by western blot analysis. As illustrated in Fig. 4, expression of N-cadherin and ERK-1 protein increased significantly, and expression of E-cadherin decreased significantly (p < 0.05) (Fig. 4).
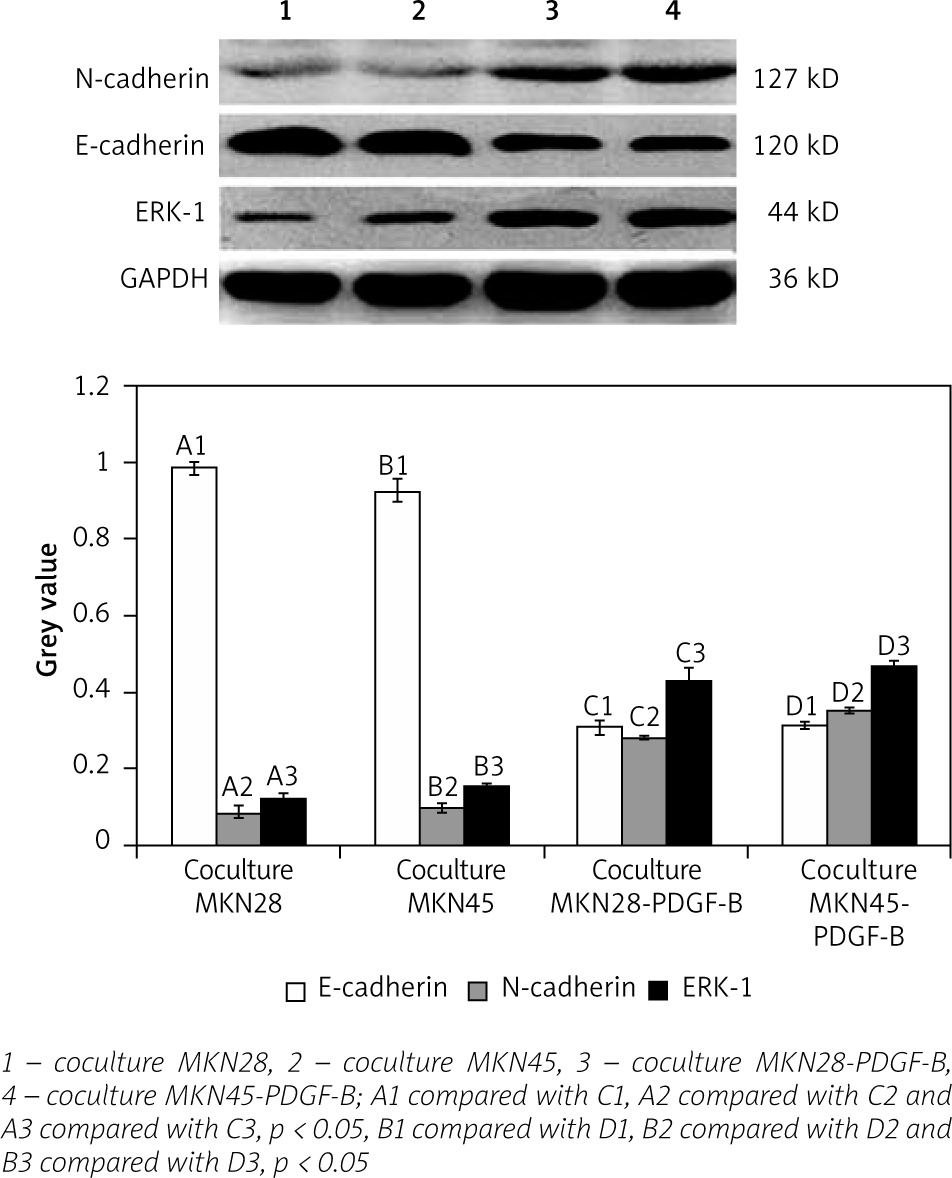
Expression of E-cadherin, N-cadherin, and ERK-1 in cocultured platelet-derived growth factor-B overexpression MKN28 and MKN45 gastric carcinoma cells after adding MAPK inhibitor
For further detection of the role of the MAPK/ERK pathway in EMT induced by activation of PDGF-B signalling, MAPK inhibitors were added into cocultured PDGF-B overexpression gastric carcinoma cells (MKN28 and MKN45). Then, expressions of E-cadherin, N-cadherin, and ERK-1 protein in cocultured PDGF-B overexpression MKN28 and MKN45 cells were detected by western blot analysis. As illustrated in Fig. 5, expressions of N-cadherin and ERK-1 protein were decreased significantly by MAPK inhibitor, and expressions of E-cadherin were increased significantly by MAPK inhibitor (p < 0.05) (Fig. 5).
Discussion
Metastasis is a common clinical finding in many human cancers, and it is the primary cause of death for most cancer patients [19, 20]. For many years, scientists have tried to understand the mechanisms of tumour metastasis, but the results are unsatisfactory. Although the exact mechanisms of metastasis are still unknown, it is widely accepted that EMT is of vital importance in the process of tumour metastasis [5–7]. PDGF-B signalling is one of the important growth factors that were demonstrated to be concerned with EMT [10, 21]. Also, PDGF-B signalling was highly expressed in gastric cancer cells, and its specific receptor PDGFR-b was highly expressed in tumour stromal cells of gastric cancer [22, 23]. Hence, we inferred that activation of PDGF-B signalling might induce EMT to promote tumour metastasis in gastric cancer. EMT is a complex molecular and cellular process and is regulated by varied of biology factors [24]. EMT is a coordinated molecular and cellular change defined as a reduction in cell-cell adhesion, apical-basolateral polarity, and epithelial markers, as well as an acquisition of motility, spindle-cell shape, and mesenchymal markers [6]. Many EMT-associated markers, such as epithelial specific markers (i.e. E-cadherin and cytokeratin) [25] and mesenchymal specific markers (i.e. N-cadherin and vimentin) [26, 27], were used for the detection of EMT. In our study, we tried to verify that activation of PDGF-B signalling might promote EMT in gastric cancer cells and tried to explore its mechanisms.
In our study, we found that PDGF-B overexpression MKN28 and MKN45 gastric carcinoma cells follow EMT just after being cocultured with cancer-associated PDGFR-β-positive fibroblast. The result showed the indispensable role of cancer-associated PDGFR-b-positive fibroblast in the EMT of gastric carcinoma cells induced by activation of PDGF-B signalling. Cancer-associated fibroblasts, the same as extracellular matrix (ECM), myofibroblasts, immune cells, and soluble factors, were an important component of tumour microenvironment [28, 29] and were widely involved in the process of EMT in many cancer cells [30–32]. Also, many studies showed that interaction among cancer cells in the tumour microenvironment can induce EMT by many mediators such as growth factors, cytokines, and ECM proteins [33, 34]. Hence, we inferred that PDGF-B, an important growth factor, could induce EMT in gastric carcinoma cells through interaction with the tumour microenvironment. The potential mechanisms might be that activation of PDGF-B signalling could induce the variations of tumour stromal cells so as to change the tumour microenvironment, such as hypoxia, acid environment, hypermetabolism, and so on [35, 36]. However, the exact mechanism(s) of how the tumour microenvironment regulates activation of PDGF-B signalling and promotes EMT is/are still not yet determined.
Also, for further exploration of the potential mechanism of how activation of PDGF-B signalling promotes EMT, MAPK/ERK signalling, a prominent tumour metastasis-related downstream pathway was detected. MAPK/ERK signalling [37] could be activated by many kinds of growth factors to promote EMT of tumour cells [38–40]. Moreover, many studies have documented that PDGF-B plays a significant role in gastric carcinoma metastasis [11, 22]. Hence, we sought to investigate whether the effects of PDGF-B signalling activation on metastasis of gastric carcinoma was mediated through activation of MAPK/ERK signalling. Significantly, activation of PDGF-B signalling increased the expression of ERK-1 and N-cadherin protein and decreased the expression of E-cadherin protein in MKN28 and MKN45 gastric carcinoma cells. Also, when PDGF-B signalling activation MKN28 and MKN45 gastric carcinoma cells were treated with MAPK inhibitor, the expression of N-cadherin protein was decreased and the expression of E-cadherin protein was increased. We could infer that activation of PDGF-B signalling induces EMT of gastric carcinoma cells at least partially through the activation of MAPK/ERK signalling. It is well known that MAPK inhibitor might inhibit the activation of ERK-1, but it was quite strange that when MAPK inhibitor was added, the expression of ERK-1 protein was decreased. We speculated that MAPK inhibitor might inhibit the activity of PDGF-B signalling through some mechanisms [41]. First, downstream pathways of MAPK/ERK signalling might be inhibited, and negative feedbacks were transferred to PDGF-B signalling. Then, activation of PDGF-B signalling was decreased [42]. However, the exact mechanism(s) of how PDGF-B signalling regulates the MAPK/ERK signalling is/are still not yet determined.
Conclusions
We found that PDGF-B signalling can induce EMT in gastric carcinoma cells. The tumour microenvironment is imperative in the process of PDGF-B signalling inducing EMT in gastric carcinoma cells. Also, activation of the MAPK/ERK pathway, which is a downstream pathway of PDGF-B signalling, might participate in this process. Thus, blockage of PDGF-B signalling pathway may be a reasonable approach to the treatment of gastric carcinoma.






