
Introduction
Gliomas are a group of brain tumors originating from the glial cells (either astrocytic or oligodendroglial). Their classification is based on cell biology, histology and clinical evolution. Although the current classification of brain tumors includes genetic and epigenetic abnormalities and clinico-pathological features, clinicians are still using the historical classification to define the tumor entities. Until recently, gliomas were divided into low-grade gliomas (LGGs) (grade I–II) and high-grade gliomas (HGGs) (grade III and IV), according to the 2007 report of the WHO classification [1]. The WHO presented in 2016 a major restructuring of the embryonal central nervous system tumors, by incorporating new entities defined both by molecular and histological features, including IDH-wildtype glioblastoma, IDH-mutant glioblastoma; H3 K27M-mutant diffuse midline glioma; RELA fusion-positive ependymoma; WNT- and SHH-activated medulloblastoma; and C19MC-altered multilayered rosettes embryonal tumor [2]. There are several studies showing that the molecular background of the discussed entity is very complex [3, 4]. Basically, because of the discrepancies in the clinical evolution of tumors with different molecular background, this classification is a problematic issue. HGGs are the most aggressive brain tumors among gliomas. The median survival of patients diagnosed with HGGs is only 14.6 months [5]. HGGs include anaplastic astrocytoma (AA), anaplastic oligodendroglioma (AO) and glioblastoma (GB). The origin of these tumors is in the supporting neuroglial cells of the central nervous system. The most aggressive of these primary brain tumors are GB. It is obvious that GB individuals require special attention and care, mainly because all HGGs can be debilitating, causing physical and cognitive impairment, epileptic seizures, depression and personality changes. In the last years, specialists have focused their energy on providing new therapies for these patients, in order to improve their lifestyle and survival. In spite of the efforts made until now, the standard of care of newly diagnosed GB remains surgery (maximal safe resection) followed by radiotherapy and adjuvant chemotherapy [6–8]. The adjuvant temozolomide associated with radiotherapy has improved the median survival, which was only 12.1 months. However, several problems linked to resistance towards chemotherapy or radiotherapy need to be solved. Microenvironment, cellular morphology and genetic characteristics are a few of the aspects to which cancer cells can adapt in order to survive, leading to drug resistance [9]. In the light of these data, it is obvious that the decisions regarding the treatment must be taken on an individual basis. In recent years, specialists focused on targeted molecular therapies. Known to be involved in cancer development and therapy, receptor tyrosine kinases (RTKs) are of particular importance [10–13].
New chemotherapeutic strategies in GB treatment are frequently proposed, but drug development and registration are consuming increased financial resources and time. Therefore, drug re-purposing represents a new pipeline for the pharmaceutical industry to find new uses in oncology for already existing non-cancer drugs. In this review we focused on some therapeutic agents that are at different stages of research or in clinical phases endowed with the potential to become re-purposing candidates for GB treatment.
Receptor tyrosine kinases
The molecular structure of RTKs includes a ligand-binding region in the extracellular domain, a single trans-membrane helix, and a cytoplasmic region. The cytoplasmic region includes the protein tyrosine kinase (TK) domain and the additional carboxy terminal and juxtamembrane regulatory regions. In the human proteome there are 58 currently known RTKs divided into 20 families [14]. RTKs are involved in regulating proliferation, differentiation, cell survival, metabolism, cell migration, and cell cycle control [15]. In 1990, Ulrich and Schlessinger demonstrated that the activation of RTKs by growth factor binding results in the dimerization and/or oligomerization of the receptor [16]. Actually, the dimerization can be ligand- or receptor-mediated, or both receptor- and ligand-mediated [17]. The dimerization of the extracellular regions of RTKs leads to activation of the intracellular tyrosine kinase domain. These changes lead to the release of the cis-autoinhibition while the trans-autophosphorylation is enabled and the tyrosine kinase domain becomes active [18]. Also, the autophosphorylation of RTKs results in the recruitment of downstream signaling proteins which contain Src homology-2 or phosphotyrosine-binding domains. By binding these domains to specific phosphotyrosine residues, the cellular signaling pathway is activated [19]
Another response to the activation of RTKs is the down-regulation of the receptor. The result of this process is the degradation of the ligand and of the receptor [20]. Also, it is known that there is a connection between the function of protein kinases and ubiquitylation, which is very important in some critical events involved in cell signaling such as regulation of protein degradation, processing and cellular trafficking [21]. The mutations or aberrant activation of the intracellular signaling pathways of RTKs are linked to a series of diseases including cancer, arteriosclerosis, diabetes, and angiogenesis. Therefore, in recent years serious efforts were made to develop molecular targeting drugs able to fight the RTK aberrations. The most studied molecular targets are epidermal growth factor receptor (EGFR), insulin growth factor receptor (IGFR), vascular endothelial growth factor receptor (VEGFR), platelet–derived growth factor receptor (PDGFR) and fibroblast growth factor receptor (FGFR) [22, 23].
Receptor tyrosine kinase inhibitors for glioblastoma treatment
In accordance with the National Cancer Institute (NCI) and the US Food and Drug Administration (FDA), targeted therapies are a set of drugs capable of blocking molecular targets involved in growth, spread and tumor progression [24]. Being designed to interact only with the molecular target, such therapies spare the normal cells. Also, being able to inhibit tumor cell proliferation, they are cytotoxic. Therefore, they can be considered instruments of precision medicine. Small molecule therapies against RTKs are among these targeted therapies. Currently, some of them are used in preclinical studies, while others have already been approved for clinical trials or for clinical use in tumor treatment including HGGs, as mentioned in Figure 1.
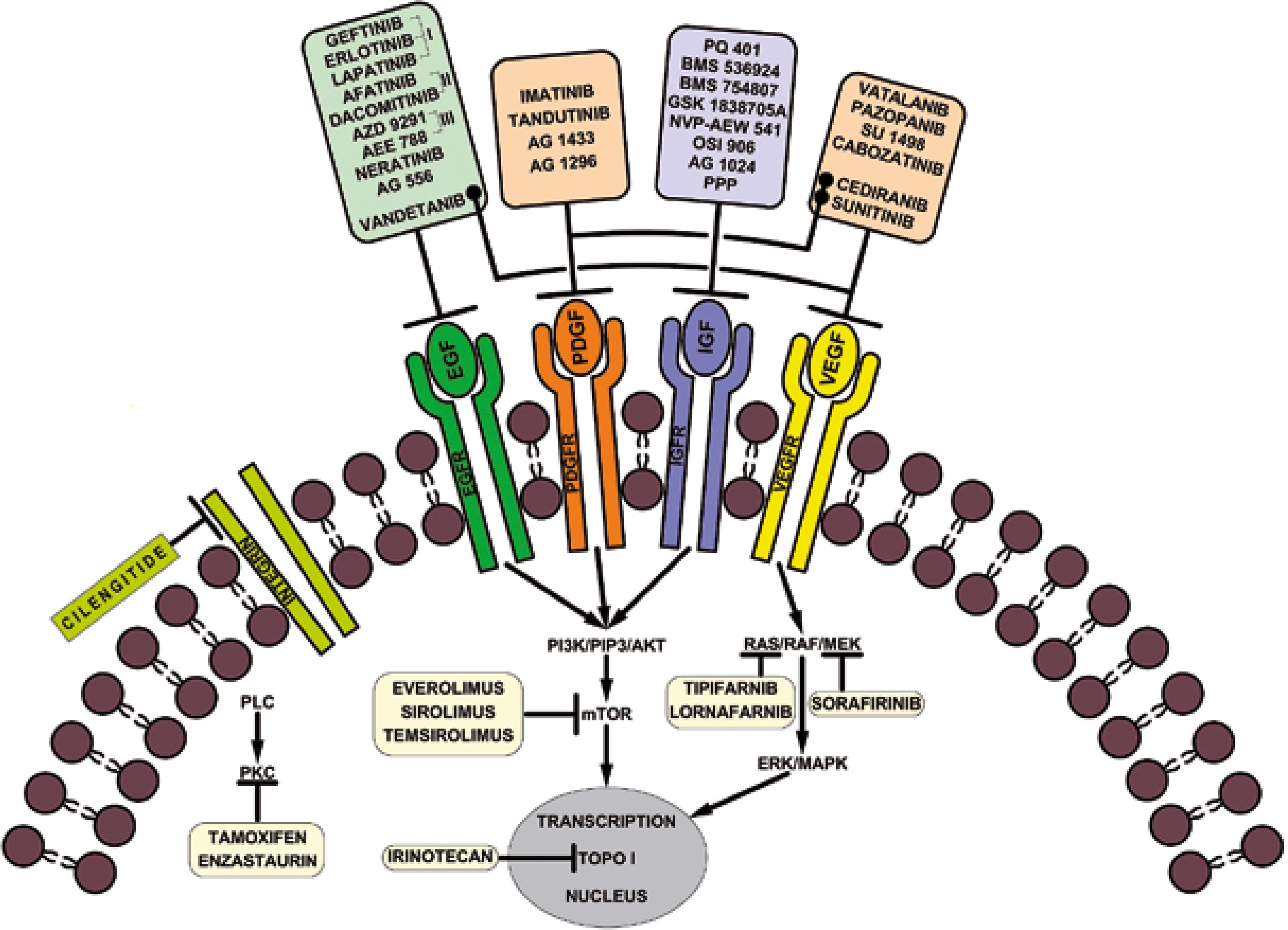
The most relevant RTK inhibitory drugs used in cancer therapy are briefly presented in Table 1.
Table 1
Small molecule receptor tyrosine kinase inhibitors used in cancer therapy
| Target | Molecules | Observations |
|---|---|---|
| EGFR | 1st generation inhibitors: Gefitinib Erlotinib Lapatanib | They showed promising results in preclinical studies, but with mixed results in clinical trials [22–25] |
| 2nd generation inhibitors: Afatinib Dacomitinib | Both drugs were approved by the FDA Afatinib had limited activity in combination with temozolomide [33] | |
| 3rd generation inhibitors: AZD 9291 AEE 788 | AZD 9291 proved to have better activity and selectivity than the previous inhibitors The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma [36] AEE 788 also inhibits VEGFR [38] | |
| Others: Vandetanib Neratinib AG556 | Vandetanib also inhibits VEGFR [39] AG556 had promising results when used in combination with radiotherapy [43] | |
| PDGFR | Imatinib mesylate Tandutinib AG 1433 AG 1296 | Imatinib showed no significant changes in the HGGs and especially GBM tumor growth [46] Better results were obtained in combination with hydroxyurea [47] Tandutinib had little effect [49] AG 1433 and AG 1296 used alone are rather effective [50, 51] |
| IGF-R | PQ 401 Picropodophyllin BMS 536924 BMS 754807 NVP-AEW 541 OSI 906 AG 1024 | PQ 401, BMS 536924 and picropodophyllin suppressed the growth and migration of GBM cells GSK 1838705A and NVP-AEW541 induced apoptosis [63–67] OSI 906 and BMS 754807 had good results in vitro AG1024 had rather modest inhibition activity alone or in combination with radiotherapy [68] |
| VEGFR | Vatalanib Pazopanib Sunitinib Cediranib Thalidomide Cabozantinib SU 1498 | Vatalanib enhances the antiangiogenic activity [54] Disappointing results were obtained for pazopanib in combination with lapatinib [57] No promising activity for GBM patients treated with sunitinib [58] Cediranib is an inhibitor of VEGFR, PDGFR, and c-kit [59] Thalidomide had a good effect as palliative drug in advanced secondary glioblastoma [60] Cabozantinib had good results both in vitro and in clinical trials [61, 62] SU1498 had a limited anti-tumor activity [51] |
[i] EGFR – epidermal growth factor receptor, PDGFR – platelet-derived growth factor receptor, IGF-R – insulin-like growth factor receptor, VEGFR – vascular endothelial growth factor receptor, FDA – Food and Drug Administration, ERK – extracellular signal-regulated kinases, HGGs – high-grade gliomas, GBs – glioblastomas, c-kit – transmembrane tyrosine kinase receptor
Because EGFR is overexpressed in about 60% of GBs, small molecule EGFR inhibitors were developed [25]. Among the first small molecule inhibitors against EGFR preclinically tested are gefitinib (Iressa; ZD1839), erlotinib (Tarceva; OSI-774), and lapatinib (Tykerb/Tyverb; GW572016). These inhibitors showed promising results in preclinical studies [26, 27]. However, the results were rather mixed in clinical trials. Gefitinib alone or in association with radiotherapy proved to have only a minimal response in patients diagnosed with GBs, although the drug was well tolerated [28, 29]. However, in 2005, Franceschi et al. proved in a phase II study of the Grupo Italiano Cooperativo di Neuro-Oncologia (GICNO) that the drug could be more efficient as a second line treatment for patients with HGGs [30]. In recent years, clinical studies proved to have similar results [31].
Similar results were obtained with erlotinib [32, 33]. Even in more recent years the drug showed only minimal benefits [34]. Lapatanib, another first generation EGFR inhibitor, also had only limited results in clinical trials either alone or in combination with temozolomide [35, 36]. Because of these rather poor results, a second generation of EGFR inhibitors was designed to inhibit the EGFR. Among them, afatinib and dacomitinib were approved by the FDA. In 2015, a phase I/phase II study regarding afatinib alone or in combination with temozolomide proved that the drug was safe but with limited activity [37]. Also, single-agent dacomitinib proved to have limited activity in a phase II clinical trial in recurrent glioblastoma patients with EGFR amplification [38], following preclinical studies with good results [39]. The third generation of EGFR inhibitors is nowadays being tested pre-clinically, but also in clinical trials. AZD9291 demonstrated to be efficient both in vitro and in vivo GB models. This drug has better activity and selectivity than the previous inhibitors. The drug has a better capacity to inhibit proliferation and prolongs the survival of GB cells [40]. Since 2018, the drug is being tested in a phase I/phase II clinical trial [41]. Another EGFR/Erb inhibitor is AEE788. The drug also inhibits VEGFR. It was tested in a phase I clinical trial developed for patients diagnosed with recurrent GB. The results were disappointing due to the toxicity and minimal activity of the inhibitor [42]. Neratinib is another inhibitor of EGFRs investigated in clinical trials for GB patients [43].
In the last years, we also investigated a number of small molecule EGFR inhibitors as potential targeted therapy on HGG cell lines. In 2018 we investigated the effect of tyrphostin AG556 (an EGFR inhibitor) on 11 and 15 HGG cells. Currently used as monotherapy, the inhibitor had only modest results. However, when combined with radiotherapy, the inhibitor induced radiosensitivity in 11 HGG cells [44]. This proved once again that HGG cells are able to develop resistance to therapies. The capacity of these cells to synthesize constitutive active receptors makes the targeted therapies ineffective.
PDGFR is another family of receptor tyrosine kinases that is overexpressed in HGGs, especially in GBs [45]. PDGFRA is amplified in about 15% of GBs [46]. This explains the efforts made to discover and test new small molecule inhibitors to target this receptor. Currently, many inhibitors are undergoing in vitro and in vivo preclinical tests and some of them are already approved for clinical trials. Imatinib mesylate (Gleevec/ST1571) is a small molecule inhibitor which has inhibitory effects on PDGFR. Although the inhibitor proved to have good effects for other malignancies, in the case of HGGs and especially GBs, imatinib mesylate showed no significant changes in the tumor growth. The drug failed the clinical trials and the patient survival remained unchanged [47]. Because of these facts, the inhibitor was next tested in combination with hydroxyurea, another classical chemotherapeutic drug. The clinical trial concluded that the combination had no benefit when compared to the single treatment with hydroxyurea [48]. In the last years, in vitro studies on GB cells proved that imatinib mesylate increases the migration and invasion of GB cells, a fact that explains the anterior failures of the drug [49]. Tandutinib, a PDGFRB inhibitor, was also tested in clinical trials in patients with recurrent GB. The drug had little effect [50]. Even since 2008 we have been interested to test the effect of AG1433, which is also an in vitro PDGFR inhibitor in several HGG cell lines (8, 18, and 38). The results were promising [51].
In 2015 we also tested the effect of the same inhibitor, AG1433, on GB9B cells in vitro. The cytotoxic effect of the drug was rather modest [52]. In the same period, another tyrphostin, AG-1296, had good effects on GB cells both in vitro and in vivo [53].
In 2019, we reported the effect of AG1433 alone and in combination with radiotherapy on 11 and 15 HGG cell lines. We found that although the use of the inhibitor alone was rather effective, the association with radiation therapy was not more effective when compared with the single treatment [54].
VEGFR is another target for glioblastoma patients. Vatalanib (PTK787) is an inhibitor of VEGFR2, PDGFR and c-kit which had little effect on GB patients alone or in combination with other chemotherapeutics or radiotherapy. However, the drug seemed to enhance the antiangiogenic activity [55]. Sorafenib is another small molecule inhibitor of VEGFR with a small effect on GB when used in combination with temsirolimus. It is in a phase II clinical study [56]. Tivozanib is a small molecule inhibitor of angiogenesis with good anti-angiogenic effects on GB. However, the drug was not able to change the volume of the tumors [57]. Pazopanib was also tested in clinical trials in combinations with lapatinib. The results were rather disappointing [58] In 2013, Batchelor et al. reported that cediranib, a small molecule inhibitor of VEGFR, PDGFR, and c-kit, showed a small effect on the neurological status of the patients but did not improve the progression of the disease or the survival of the GB patients [59]. Another anti-angiogenic agent which proved good effects on GB patients is thalidomide. The drug had a good effect when used as a palliative drug for patients with advanced secondary GB [60]. SU1498 is a VEGFR inhibitor that proved to have a cytotoxic effect on GB9B cells. However, its anti-tumor activity was rather limited [52].
YKL-40, a mesenchymal marker known as human cartilage glycoprotein-39 or chitinase-like protein 1, seems to have a key role in the motility and migrating features of glioma stem like cells and in their differentiation into endothelial cells, involved in angiogenesis [61]. It was proven that YKL-40 upregulates VEGF expression, and tumor vasculogenesis induced by YKL-40 is partially dependent on VEGF [62]; therefore therapies targeting YKL-40 may have potential benefit in GB treatment.
IGF-1R is another receptor tyrosine kinase that proved to be an interesting target for GB treatment. In the last years, a number of small molecule inhibitors against IGF-1R have been tested on GB cells in vitro and in vivo. Among them, PQ401, BMS-536924 or PPP (picropodophyllin/AXL1717) proved to be able to suppress the growth and migration of GB cells, while GSK1838705A or NVP-AEW541 induced apoptosis either alone or in association with other chemotherapeutic drugs [63–67]. Also, inhibitors such as OSI-906 and BMS-754807 proved good results in vitro on GB cells [68]. Our group studied since 2007 the capability of tyrphostin AG1024 to inhibit IGFR on a series of HGG cells lines. First we studied the 18 and 38 HGG cell lines [69]. In the next year, we added some other HGG cell lines: MO59J, MO59K, and 8. The activity of the inhibitor was rather modest. Similar results were obtained when combining the inhibitor with ionizing radiation [51].
Somatic mutations of FGFR are rare in GB, but there are studies suggesting that modifying FGFR signaling influences glioblastoma progression and patient survival [70]. Small molecules which inhibit the FGFR tyrosine kinases are currently being studied, emphasizing the therapeutic potential of this signaling pathway [71]. Some small-molecule inhibitors such as lenvatinib, ponatinib, dovitinib and brivanib, also target other RTKs, while others are FGFR selective, such as PD173074, BGJ398, AZ4547, and JNJ-493 [72].
In a recent study, a large-scale shRNA screen was used to identify FGFR signaling as a target in pediatric glioma, proving that dovitinib, ponatinib, AZ4547, and PD173074 better reduce the growth of glioma cells in vitro than temozolomide [73].
In December 2019, a trial involving BGJ398 in patients with recurrent glioblastoma was completed, but so far, no results have been published [74].
A phase I/II trial involving TAS-120 is currently recruiting patients with advanced solid tumors, with and without FGF/FGFR-related abnormalities [75].
Rapamycin (sirolimus) has been identified to inhibit the mTOR and, specially, the mTORC1 complex [76]. Rapamycin derivatives (temsirolimus, everolimus and ridaforolimus), also named rapalogues, have been synthesized. At present, they are gaining considerable interest. By using clinicaltrials.gov lists regarding sirolimus/everolimus/temsirolimus treatment in GB patients, we found that 7 clinical trials were recruiting in 2019 [77].
The multitargeted approaches may represent a method for effective selection of resistant tumor subclones. Vandetanib is a multitargeted tyrosine kinase inhibitor (VEGFR, EGFR) that was studied in clinical trials. The drug was well enough tolerated but the antitumor effects were limited [78]. In 2015 another phase I clinical study determined that the co- administration of vandetanib in association with sirolimus is safe for patients with recurrent GB [79]. Also, in another clinical trial vandetanib proved to be safe in association with standard chemotherapy in newly diagnosed GB patients [80]. Two other multitarget small molecule inhibitors which target VEGFR and other receptors are XL-184 (cabozantinib) and PD173074. Cabozantinib had good results both in vitro and in clinical trials, and PD173074 had good results in vitro [81, 82]. Sunitinib is a multiple kinase inhibitor of VEGFR, PDGFR, FLT1, FLT1/KDR, FLT3, and RET kinases with no promising activity for GB patients [83].
In conclusion, targeted therapy against receptor tyrosine kinases represents a hope for GB patients. However, the efforts made by specialists should also be focused on fighting against resistance to therapy, to discover drugs able to pass the blood brain barrier, to use multi-targeted therapies, but also to discover and use biomarkers that can predict the outcome of therapies.
Antibody therapies targeting the RTKs’ extracellular domain
Apart from the kinase domain, the extracellular domain of RTKs may represent a viable target by using antibody therapies as antagonists. Because of their large size, they do not freely cross the blood–brain barrier (BBB); therefore engineered antibodies (such as directed antibodies with transferrin receptor optimized binding) must be used to enable them to access the GB tumors. Also, to bypass the BBB, alternative antibodies can be delivered inside the brain using Ommaya reservoirs [22].
Cetuximab is a monoclonal EGFR targeting antibody used for GB treatment. It prevents RTK activation by targeting the extracellular domain of EGFR [84]. Its activity was minimal in phase II clinical trials on recurrent GB patients [85]. Also, onartuzumab was used to inhibit the tumor growth of orthotopic U87 GBM xenograft [86]. Dalotuzumab (MK-0646 or H7C10/F50035) is a humanized monoclonal IGF-1R antibody shown to induce apoptosis and to reduce cell proliferation [87].
RTK drug resistance in glioblastoma patients
The resistance to RTK drugs has many causes. Usually, monotherapies yield minor results, mostly because of the functionally redundant pathways. Due to the fact that intracellular signal redundancy is the main cause of therapeutic failure by using a single inhibitor, concurrent blocking of multiple receptors or of an RTK inhibitor together with radio-, chemo or immunotherapy is an applicable strategy. Recent preclinical and clinical studies suggest the need for concomitant inhibition of multiple RTKs or for inhibition of their common downstream signaling. Hence, there has been a growing interest in testing the inhibitors of PI3K, AKT and the TORC1/2 complexes. A multitarget treatment may be a good solution when certain subclones of the tumor become resistant to single treatment by creating mutations; therefore an option to overcome resistance is to act selectively on these mutations.
There are two types of approaches mentioned in the literature: the vertical inhibition approach in which the molecular targets are part of the same cellular signaling axis, and the horizontal inhibition approach where the multitarget ligand is involved in distinguished nodes of different pathways [88]. These approaches are achieved by using co-administration of drugs (Akt/mTOR, MDM2/mTOR, PI3K/CDK inhibitors) or by using multi-target ligands (PDK1/Aurora A, PDK1/CHK1, Akt/p70S6K, EGFR/PKC inhibitors) [89]. For example, a study performed by Graves-Deal et al. showed that the multi-RTK inhibition strategy managed to overcome both de novo and acquired resistance to EGFR therapies. The efficiency of multiple EGFR-targeted antibodies (panitumumab, cetuximab, and MM-151) could be enhanced by adding small molecule RTK inhibitors (crizotinib, cabozantinib, and BMS-777607). Also, by adding crizotinib, resistance to cetuximab in nude mice xenografts was overcome [90]. This strategy could also be applied for GB treatment. Wei et al. performed a study on patient-derived glioblastoma xenografts grown in mice. The results showed that copy number variations and mutations did not correlate with drug resistance, but increased heterogeneity and activation of the ERK and SRC kinases in drug-resistant tumors. The tumor growth was prevented by combining the different pathway inhibitors (mTOR, ERK, SRC) in mTOR inhibitor-resistant GB mice. The rewiring events were detected a few days after the beginning of the treatment, at the single-cell level [91].
Another option to overcome the RTK inhibitors resistance is to use blockers for the apoptosis inhibitors. In a study performed by Ziegler et al., in vitro inhibition of PDGFR in human GB cells started the apoptosis intrinsic pathway, but caspase activation could be blocked by inhibiting the apoptosis proteins. Therefore, concomitant inhibition of apoptosis proteins may overcome the resistance to RTK inhibitors, improving treatment outcomes [92].
In conclusion, the concurrent inhibition of different cellular pathways is a new promising strategy that is attempting to overcome the onset of chemoresistance. These strategies may involve the combination of multiple selective inhibitors blocking different targets in the same pathway, the concurrent blockade of key proteins of the signaling pathways, or the multidirectional inhibition of specific oncoproteins. All these approaches represent a valid strategy in GBM therapy, especially when the patient genetic pattern is the target.
Repurposed drugs with potential use in glioblastoma therapy
There are many innovative chemotherapeutic strategies developed in GB treatment, but nowadays regulations concerning drug development and registration require a long time and increased financial resources. The pharmaceutical industry is trying various other pathways in order to put drugs faster on the market. Besides that, it is known that physicians may prescribe “off-label” drugs, though this represents a controversial practice in some fields (pediatrics, oncology) [93]. One strategy applied is drug repositioning, also known as drug re-purposing. It is a pharmaceutical strategy applied in oncology and other areas based on finding new indications for already approved drugs, in order to treat off-label diseases [94].
The reason for using drug re-purposing is due to the ability of small molecule agents to target distinct cellular proteins. Thus, the same molecule can be used to target multiple pathways involved in malignant diseases that are usually considered to be unrelated (polypharmacology) [95].
Because it skips many phases [96], as can be seen in Figure 2, this strategy has many advantages.
Fig. 2
De novo and re-purposing drug development phases. Unlike the drugs that follow the conventional pathway to the pharmaceutical market, re-purposing candidates shorten the time needed to market by omitting some initial steps, which go directly into the clinical study phases (FDA – Food and Drug Administration. I, II, III – stages in the clinical development [clinical phases])
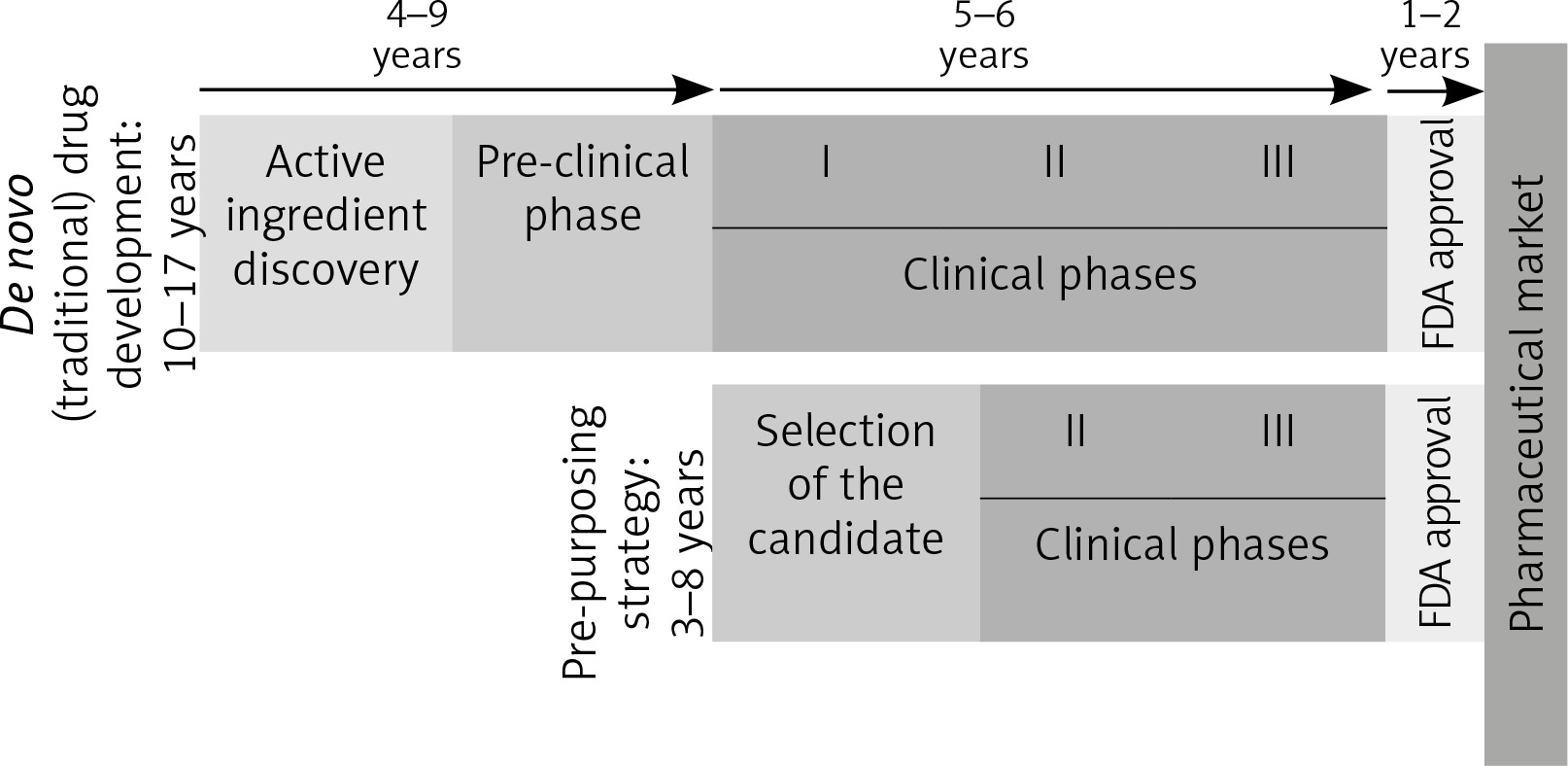
The molecules already in use have well-known pharmacological data, a fact that shortens the period for approval, but also the final price. Furthermore, most of these drugs are generics, so their cost of production is lower than for the patented drugs [97].
However, key obstacles must be mentioned such as registration, reimbursement and implementation of the re-purposed drugs. For example, in Europe only the holder of the marketing authorization can apply for the extension of a marketing authorization [98]. Also, non-commercial organizations usually lack resources required to finish and maintain the marketing authorization. There is still a doubt regarding the necessity of large randomized controlled trials to confirm the efficiency of a re-purposed agent and the use of the authorization dossiers’ safety data, in order to overleap phase I studies [99].
For example, out of 44 off-label recommendations listed in the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines), only 14 were approved by the FDA and/or are subjects of randomized controlled trials [100].
Concerning central nervous system (CNS) therapy, only the FDA approved agents with ability to cross the blood brain barrier (BBB) may become re-purposing candidates [95]. By using the PubMed database published between 2010 and 2019 and the site clinicaltrials.gov, we gathered a list of agents that have re-purposing potential for GB treatment. In this short review, we indicated the various approaches used to repurpose drugs in GB therapy and we also highlighted their limitations.
Some of the mechanisms involved in GB therapy were completely elucidated, but many still remain unclear. In Figure 3, we illustrated the plethora of drugs with repurposing potential in GB therapy, but the number of studies in this field is significantly higher [94, 101–126].
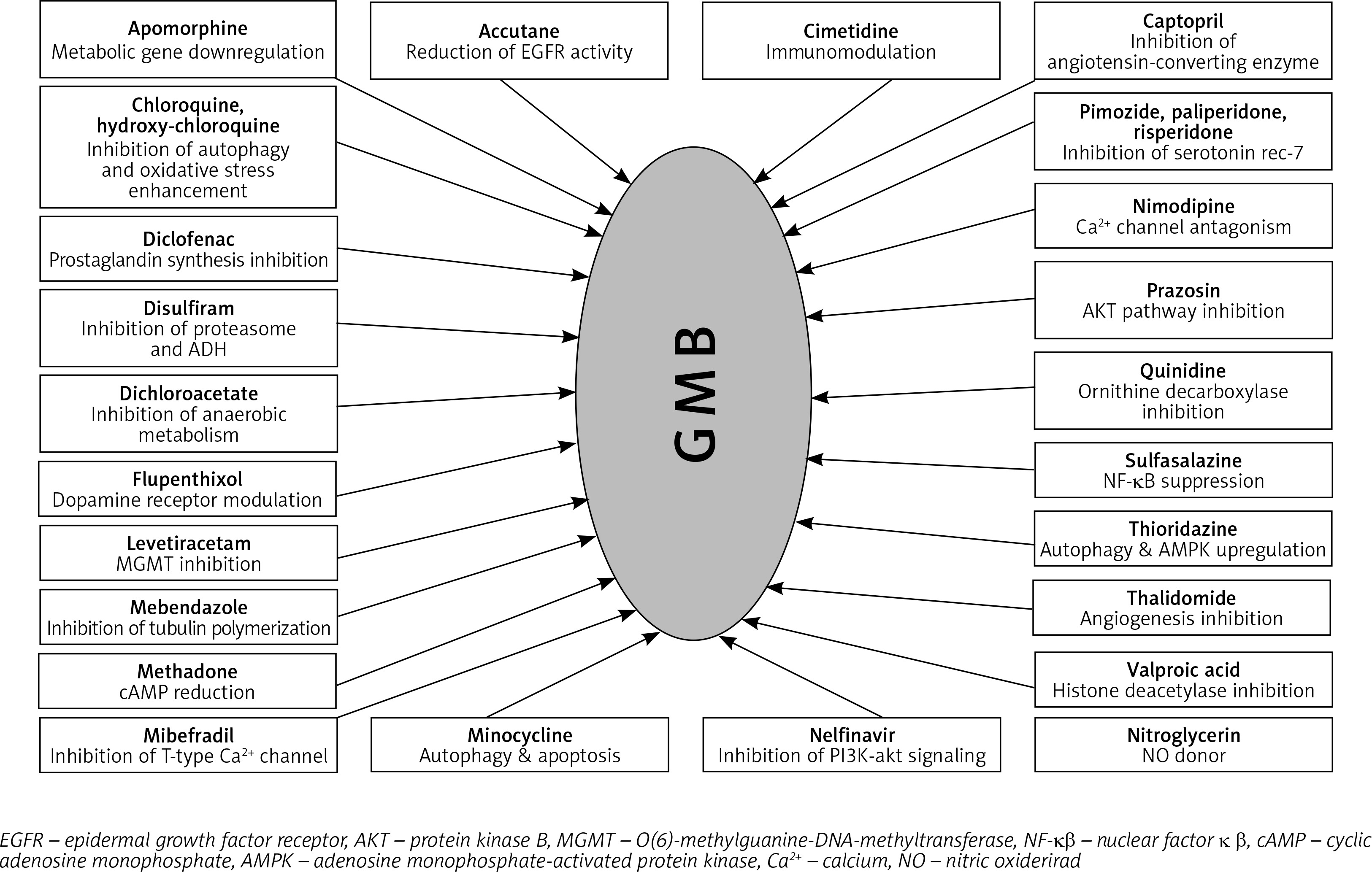
In this review, we focused on the following drugs that were tested as re-purposing candidates: CNS drugs (chlorpromazine, pimozide, fananserin, trifluoperazine, thioridazine, imipramine, valproate, propentofylline), antimalarial drugs (chloroquine, mefloquine), antidiabetics (metformin), disulfiram, lonidamine, rapamycin, temsirolimus, everolimus and ridaforolimus.
Several studies investigated the properties of some FDA-approved psychotropic molecules to inhibit the proliferation and migration of GB cells [109, 127].
For example, chlorpromazine is a specific and potent inhibitor of the kinesin KSP/Eg5 leading to mitotic arrest and defective, monopolar spindles [128]. It is also involved in autophagic cell death due to inhibition of the AKT/mTOR signal transduction axis in human glioma cells [129]. Antipsychotic drugs such as chlorpromazine and pimozide were tested on glioblastoma cells and they showed tumor suppressing ability [130].
Fananserin, a dopamine receptor D4 (DRD4) inhibitor, selectively induced autophagy in GB stem cells [131].
Trifluoperazine, a dopamine receptor D2 antagonist, inhibits both growth and proliferation of GB cells in a dose-dependent manner [132].
A recent study showed that thioridazine inhibits autophagy and sensitizes glioblastoma cells to temozolomide, inhibiting tumor growth in vivo and increasing survival in tumor-bearing animals [133].
It has been recently observed that antidepressants, especially imipramine and amitriptyline, can downregulate the “stemness genes” Sox1, Sox2, Ki67, Nestin, and CD44 after tricyclic antidepressant treatment. They also hypothesized that these compounds can affect tumor plasticity and immunity by influencing immune cells, reactive oxygen species and pro/anti-inflammatory cytokines [134].
Valproic acid is an anti-epileptic agent which acts by blocking sodium channels, GABA transaminase, and calcium channels [135]. Valproate is prescribed in epilepsy, migraines and acute manic episodes [136]. It was shown to have anticancer effect in glioblastoma, by reducing PON2 expression, which increases ROS production and triggers Bim output that inhibits malignant progression through the cascade PON2-Bim [137]. Furthermore, valproic acid induced autophagy through the ERK1/2 pathway which led to glioma cell death. By using the combination of valproic acid and temozolomide or rapamycin, autophagy was enhanced both in vivo and in vitro [138]. Valproic acid is also studied in different drug combinations, to increase the treatment efficiency of GB [139]. Between 2018 and 2021, valproic acid is in a phase 4 clinical trial for glioma patients with their first seizure [140].
Also, the neuroprotective drug propentofylline tested for Alzheimer’s disease and vascular dementia [141] was proven to target TROY, a receptor involved in the tumor necrosis factor receptor (TNFR) microglial signaling pathway [142].
Furthermore, the antimalarial agent chloroquine improved chemo-radiation treatment in GB [143], making it suitable as a re-purposing candidate [144]. Briefly, chloroquine (alone or in combination with temozolomide) leads to accumulation of autophagic vacuoles with non-functional properties, thus inhibiting autophagy [117, 145]. Several clinical trials are currently being conducted [146].
In 2019, a phase 1 study reported unexpectedly low rates of neuropsychiatric side effects of another antimalarial agent, mefloquine, repurposed for the treatment of GB [147].
Interestingly, a possible target in GB treatment is chloride intracellular channel1 (CLIC1), known to be inhibited by the anti-diabetic biguanides [148]. Metformin is a representative of biguanides and it is the most used oral antidiabetic drug. In 2016, a set of kinases were identified as potential targets, including SGK1 and EGFR [149]. Clinical trials are currently in different stages regarding metformin in association with other drugs, in GB therapy [150].
Disulfiram is an ALDH1 inhibitor, a staminal marker for GB [151]. The activity of disulfiram increases if administered together with divalent cations (Cu gluconate). Recently, disulfiram has been reported to inhibit NF-κB [152] and methylguanine-DNA methyltransferase [153]. Eight clinical trials are currently in different stages regarding disulfiram in association with other drugs, from which 2 are completed, in GB therapy [154].
Lonidamine is a reversible inhibitor of spermatogenesis. During its clinical use in combination with other anti-cancer drugs, it exhibited promising results in brain tumors [155]. Recently, new studies show that lonidamine inhibits the lactic acid efflux mediated by the MCT proteins. In addition, lonidamine also elicits a cytotoxic autophagic response in GB cells [156].
All these agents could be re-purposed for GB treatment, but not before a better understanding of their mechanism and formulation. Also, some researchers consider that combining the drugs with re-purposing capacity may be advantageous. In 2013, the Coordinated Undermining of Survival Paths protocol (CUSP9) was developed to assess the safety of temozolomide in GB in combination with other drugs [157]. This protocol used combinations of 9 re-purposed drugs (aprepitant, minocycline, disulfiram, celecoxib, sertraline, captopril, itraconazole, ritonavir, auranofin) and low doses of temozolomide. In this monocentric trial, all patients are treated at Ulm University Hospital Germany. This clinical trial is currently in phase 2. The estimated study completion date is March 2020 [158].
Despite the efforts, many drugs have failed to be approved for re-purposing in the treatment of GB [159, 160], as can be observed in Table 2.
Table 2
Repurposing candidates that did not receive approval for glioblastoma treatment. Although they have been considered as candidates in the re-purposing process, many old active molecules have failed to complete all steps to enter the pharmaceutical market for glioblastoma treatment, being rejected at different stages of development
Conclusions
The above described drugs are not target-specific drugs, but they can represent a therapeutic option designed rather to “target” cancer cell dependencies. Because of the heterogeneity of GB, the re-purposing approach has great potential, since their combined administration, together with current therapeutic options, could target cancer cell survival mechanisms, thus providing a strategy to avoid drug resistance in GB treatment. The information presented herein highlights the necessity of extensive research to elucidate some of the unclear biological mechanisms that underly the therapeutic effects. This step is mandatory, in order to go through all stages of developing clinical trials, until drug marketing.






