
Introduction
Despite impressive worldwide progress in the field of cancer diagnosis, prevention, and treatment strategies, cancer is still one of the leading causes of death globally. This is because cancer development and progression are driven by multiple molecular alterations that arise at the genomic and epigenomic levels [1]. This multi-step event has been termed the “hallmarks of cancer”, affecting both the nuclear and mitochondrial genome.
Mitochondria have been acknowledged as extremely dynamic organelles where their biogenesis and bioenergetic functions are closely monitored and controlled by the nuclear genome [2]. Almost a century ago, a team led by Otto Warburg described the very first theoretical origin of cancer, i.e. that cancer formation might result from altered mitochondrial metabolism [3]. Since then, researchers have made remarkable progress in every aspect of the mitochondria in understanding how defective mitochondria can contribute to cancer development and progression.
Mitochondrial genome
A unique criterion about mitochondria is that they have their own double-stranded circular mitochondrial DNA genome, called mtDNA [4]. Each eukaryotic cell carries numerous copies of mtDNA. Human mtDNA is a double-stranded circular genome of 16.5 kb, encoding 13 mitochondrial polypeptides (all of the oxidative phosphorylation [OXPHOS] system subunits), 22 tRNAs, and 2 rRNAs [4]. Unlike nuclear DNA (nDNA), intervening sequences are absent and some genes almost overlap.
This circular genome also consists of approximately 1 kb sequence harbouring a non-coding regulatory region (NCR) that serves as a control region and contains conserved domains of sequences [5]. It has the promoter sequences that stimulate the polycistronic transcription, one for each mtDNA strand named as the heavy (HSP) and light (LSP) strand promoter, 3 conserved sequence block regions, the H-strand origin of replication (OH), and the termination-associated sequence (TAS). Another important element in this region is the formation of a triple-stranded structure called displacement-loop (D-loop). This structure is created by premature termination of nascent H-strand DNA synthesis at TAS, producing the 7S DNA short sequence of approximately 650 nucleotides in length [5]. At about 11,000-bp far away from NCR, there is a L-strand origin of replication (OL) found downstream of the OH site.
Both the mitochondrial tRNAs and rRNAs are involved in the synthesis of intramitochondrial proteins. The 13 polypeptides are components of the 5 complexes involved in cellular respiration. The complexes can be divided into 2 parts, complexes I–IV form the electron transport chain, while complex V participates in adenosine triphosphate (ATP) production [6].
So far, about 1500 proteins have been found inside the mitochondria, which include around 76 contribute to the OXPHOS system that are encoded by nDNA [7]. Complex II is built up solely with no mtDNA-encoded subunits, whereas the other complexes are made up from both mtDNA-encoded and nDNA-encoded subunits.
Mitochondrial DNA genome replication has several unique characteristics that are different from the replication of the nuclear genome. The synthesis is managed by various nDNA-encoded proteins. As mentioned above, the NCR region of mtDNA contains one origin of replication for the heavy strand, while a distinct origin of replication for the light strand is placed outside the NCR region. DNA polymerase gamma (polγ), the one primary enzyme characterized in mitochondria, is responsible for mtDNA synthesis as well as its repair [8]. Human polγ is a heterotrimeric composed of a catalytic subunit (polγA) and 2 identical subunits (polγB). It has been described that polγ is also a commonly reported target of oxidative damage, which can cause a decrease in mtDNA replication rates and repair abilities [9].
Mitochondrial DNA genome transcription is launched within the NCR region at the initiation points, the HSP and LSP, resulting in long polycistronic precursor RNA [10]. The major elements needed to accomplish mtDNA transcription are the mitochondrial RNA polymerase (POLRMT), mitochondrial transcription factor A, and mitochondrial transcription factor B1 or B2 (TFB1M or TFB2M) [11]. They form a complex at the mitochondrial promoter NCR to initiate transcription. For the transition of initiation to elongation, the presence of an additional protein, termed mitochondrial transcription elongation factor, is required, which interacts with POLRMT to generate a long polycistronic transcript [12]. The primary polycistronic transcripts are carried out post-transcriptionally by cleavage of the tRNAs, which produces the individual mRNA, tRNA, and rRNA molecules [13]. Transcription from HSP generates the 12 mRNAs, 14 tRNAs, and 2 rRNAs, while transcription from LSP encodes 8 tRNAs and 1 mRNA.
Mitochondrial DNA genome copy numbers typically exist in the range of 100–10,000 per somatic cell. This variability relies on each cell type and its energy demand. In most situations, all the mtDNAs within the cells of an individual are identical, which is termed homoplasmy. However, spontaneous mtDNA alterations may arise, resulting in a mixture of wild-type and mutant genotypes coexisting within a cell, a condition referred to as heteroplasmy [14]. Because mtDNA is present as multiple copies per cell, a low level of mutant copies does not cause a pathogenic phenotype. A minimum critical number of mutant mtDNAs must be present in the cell to cause mitochondrial dysfunction. A ratio of mutant/normal mtDNAs in a cell must reach a certain threshold level (~ 60–95%) before any biochemical defects and clinical manifestations become apparent [15]. Mitochondrial DNA genomes are at risk of damage due to their closeness to the sites of reactive free radicals that are produced as a by-product during ATP synthesis. This makes mtDNA 5 to 10 times more likely to be mutated than nDNA. Moreover, the absence of protective histones and paucity of repair mechanisms are also major reasons for the higher instability discovered in mtDNA when compared to nDNA.
Accumulation of alterations can lead to mitochondrial dysfunction, resulting in a further increase of oxidative damage to cellular components, including DNA, lipids, and proteins. This progressive mtDNA damage, which leads to a remarkable reduction in mtDNA copy number, is believed to contribute to aging and has also been widely implicated in various human diseases, including cancer, diabetes, and neurodegenerative diseases [16, 17].
Mitochondrial metabolism
The main role of mitochondria is to supply over 90% of the ATP needed for cell metabolism through its OXPHOS pathway [6]. In conditions lacking mitochondria and/or adequate oxygen supply, cells rely on anaerobic respiration/glycolysis to produce all the required ATP from glucose. In anaerobic glycolysis conditions, only a net of 2 ATP can be produced from one molecule of glucose. However, under aerobic conditions, mitochondrial respiration can metabolize one glucose molecule into 32 ATP molecules.
Oxidation of fatty acids, carbohydrates, and amino acids generates high-energy electron carriers such as NADH and FADH2 that eventually transport the electrons to the mitochondrial electron-transport chain. The electrons are transferred through a series of 4 respiratory complexes (complexes I, II, III, and IV) embedded in the inner membrane to their ultimate electron acceptor, oxygen. NADH and FADH2 begin the chain by delivering their electrons to complex I and complex II, respectively. These electrons are then passed along to the next complex in the chain. While the electrons move through the electron-transport chain, protons are pumped into the intermembrane space to create a mitochondrial membrane potential gradient that develops ATP at the end. The leakage of electrons at the mitochondrial electron-transport chain, mainly from complex I and complex III, contribute to the partial reduction of oxygen to generate reactive oxygen species (ROS) [18].
Reactive oxygen species is mainly generated as a by-product of mitochondrial OXPHOS and can be detoxified by the effective antioxidant defence systems with which the mitochondria are provided [19]. Normal/healthy cells firmly regulate the constant balance between the ROS productions and antioxidant pathways to maintain a good redox balance. Conversely, it is known that elevated levels of ROS accumulate within the cancer cells and their reactions can cause oxidative damage, such as nDNA or mtDNA mutations [19].
In 1924, Otto Warburg was a pioneer in the theory of altered cancer metabolism. Warburg discovered that cancer cells tend to metabolize glucose anaerobically, instead of through mitochondrial OXPHOS, even under sufficient oxygen conditions [3]. He ultimately hypothesized that these metabolic alterations are a result of a defective mitochondrial OXPHOS. Malfunctioning mitochondria in cancer cells is thought to reflect tumourigenesis because it can modulate important processes that are linked with proliferation, migration, and invasion through a deficiency in metabolism and diminished apoptosis.
Mitochondrial DNA genome mutations
Based on numerous studies that have been carried out, scientists believe that the mitochondrial genome damage is more extensive and persists longer than that of the nuclear genome. This condition can be stimulated to produce excess ROS in the mitochondria, which are generated during ATP synthesis, and which can cause massive damage to mtDNA and genetic instability. Consequently, mtDNA alterations regularly alter energy production, which is notably harmful to cells with high energy needs like muscle and nerve cells [20]. These mutations can promote abnormalities in mitochondrial function, ultimately resulting in greater ROS production.
This progressive damage in the mitochondrial genome has been implicated to play a role in aging and age-associated diseases, including cancer, neurodegeneration disorders, and diabetes and to cause a variety of human mitochondrial diseases. Mitochondrial diseases can be caused by mtDNA alterations in the types of point mutations or multiple deletions as well as mutations in genes of nuclear-encoded mitochondrial proteins [21]. The initial finding of pathogenic mtDNA mutations in human patients was reported > 30 years ago in human patients with mitochondrial myopathies and Leber’s hereditary optic neuropathy, in 2 previous studies by Holt et al. [22] and Wallace et al. [23], respectively. Diseases that result from mtDNA instability are commonly characterized by some features of neuromuscular abnormality. Point mutations, deletions, or large loss of mtDNA molecules may contribute to premature deafness, myopathy, or severe encephalomyopathy [21]. Other point mutations of mtDNA can lead to Leber’s Hereditary Optic Neuropathy, which results in degeneration of retinal ganglion cells and vision loss. In addition, mutations in nuclear genes encoded for mitochondrial OXPHOS complexes and factors, protein import system, as well as translational machinery can lead to secondary instability of the mitochondrial genome, which results in mitochondrial diseases that follow a Mendelian inheritance pattern. Until now, over 300 pathogenic mitochondrial genome alterations have been characterized that lead to complex diseases with the heterogeneity of phenotypes (http://www.mitomap.org.), and the number continues to increase.
Moreover, mtDNA mutations have also been identified in a wide variety of cancers [24, 25], since the first descriptions by Vogelstein’s team in 1998 [26], who revealed 70% of somatic mtDNA mutations in 7 of 10 human colorectal cancer cell lines. Mitochondrial DNA genome alterations in cancers commonly discovered either in the types of somatic mutations or germline polymorphisms include point mutations, deletions, insertions, mono- or dinucleotide repeats, and mtDNA copy number variations [24]. The most frequent hot spot of mtDNA somatic mutations in various cancers is located in the D-loop region, particularly at nucleotide position 303–309, a polycytidine stretch (C-tract) known as D310 [27]. The large mtDNA deletion termed mtDNA 4977bp, which occurs between nucleotides 8470 and 13,447 of the human mtDNA, is the most common deletion associated with many types of cancer [28, 29].
Some types of cancer are also often described by the presence of mitochondrial microsatellite instability (mtMSI), which is a change in repetitive sequences mainly involved in the D-loop region [30]. The alterations in homopolymeric tract length are mainly derived from replication slippage of mtDNA polymerase, which may impair mtDNA replication and transcription.
In addition to the sequence alterations in the mitochondrial genome, variations in mtDNA copy number are commonly reported in a variety of cancers. For example, some studies have reported that mtDNA content is decreased in gastric, breast, and esophageal cancer [31–33]. However, in other types of cancer, including colorectal and pancreatic cancer, an elevated mtDNA copy number has been observed [34, 35].
A clear link between mtDNA mutation accumulation with aging has been reported in numerous human genetic diseases and in a wide variety of tissues in different species [36]. In a more recent study, Arbeithuber et al. analysed the entire mitochondrial genome by using a highly precise duplex sequencing method to detect de novo mtDNA mutations in oocytes, brain, and muscle cells of mice [36]. They revealed a 2- to 3-fold increase of mutation rate in 10-month-old compared with one-month-old mice.
Mitochondrial DNA genome microsatellite instability
Microsatellites are characterized by short tandem repeats (STRs) with highly polymorphic DNA loci, comprising repetitive nucleotide sequences (~1–10 base pairs) that are randomly abundant in coding and non-coding regions of the human genome. These DNA segments are highly vulnerable to replication errors, which may lead to mutations. Microsatellites are sensitive to mutations, with mutation rates typically ranging from 10–6 to 10–2 per generation. When alterations exist within microsatellite regions resulting in deletions or insertions (indels) of these repetitive DNA sequences, this will exhibit a feature hypermutator phenotype known as microsatellite instability (MSI) [37–39].
Microsatellite instability was believed to be caused by slippage of the DNA polymerase enzyme during DNA replication, which is involved in the generation of indels within microsatellite regions [40]. Microsatellite instability was a common molecular incident and might play an essential role in the oncogenic event. If such errors occur within hotspot coding regions that possibly caused frameshift mutations, these alterations may disrupt normal cellular functions, thus leading to altered homeostasis and increased risk of cancer [40–42].
In the year 1993, Thibodeau et al. were the first to encounter MSI in relation to tumour development [43]. They revealed that 28% (25/90) of the colorectal cancer cases had MSI deletion mutations in “CA repeats” or (CA)nsequence region, which was MSI, initially known as MIN. Moreover, the remarkable discovery showed that MSI was significantly associated with the proximal colon tumours and that the patients with MSI tumours exhibited a better prognosis [43]. Since then, MSI research has greatly expanded in various human cancers, and researchers have focused extensively on MSI-disease connections to gain more insight into the basic molecular mechanism [44, 45].
Mitochondrial MSI has been described as the alteration in the length of short repetitive sequences of mtDNA between normal and tumour tissue. The occurrence of mtMSI was previously reported to be associated with mitochondrial segregation. Considering the strict nature of bottleneck segregation of mtDNA, it was proposed that mtMSI were either de novo generated in each individual or genotypically inherited [37].
As suggested, the responsible mechanism that induces mtMSI formation is similar to replication slippage in the nuclear genome. A mitochondrial MSI event was not previously reported in 8 or fewer cytosine homopolymeric sequences, (C)n tract [37]. The finding was supported by Heerdt et al. [46] and Habano et al. [47], who reported no instability occurrences in the (CA)n repeat region, but only in the (C)n tract. The replication-slippage mechanism model (also known as copy-choice recombination) proposed by Viguera et al. states that DNA polymerase pausing and dissociation appear to be involved in the induction of slippage errors [48]. In addition, Kunkel and Loeb suggested that the low fidelity of mtDNA polymerase γ was the cause of replication flaws [49].
Meanwhile, replication-slippage mistakes had dragged the mismatch repair (MMR) system and its inefficiency of repairing slippage errors [50]. The mismatch repair system plays a vital role in ensuring the fidelity of the newly synthesized DNA during replication, besides repairing DNA mutations and maintaining the stability of microsatellite regions [51]. Microsatellite instability can occur as a result of the failure of the MMR system functions when there are alterations in the MMR genes. It has been demonstrated that germline mutations or promoter hyper-methylation of MMR genes, such as MLH1, MSH2, MSH3, MSH6, and PMS2, resulted in loss of MMR function, which further caused the MSI and increased susceptibility to cancers [51].
Mitochondrial microsatellite instability formation
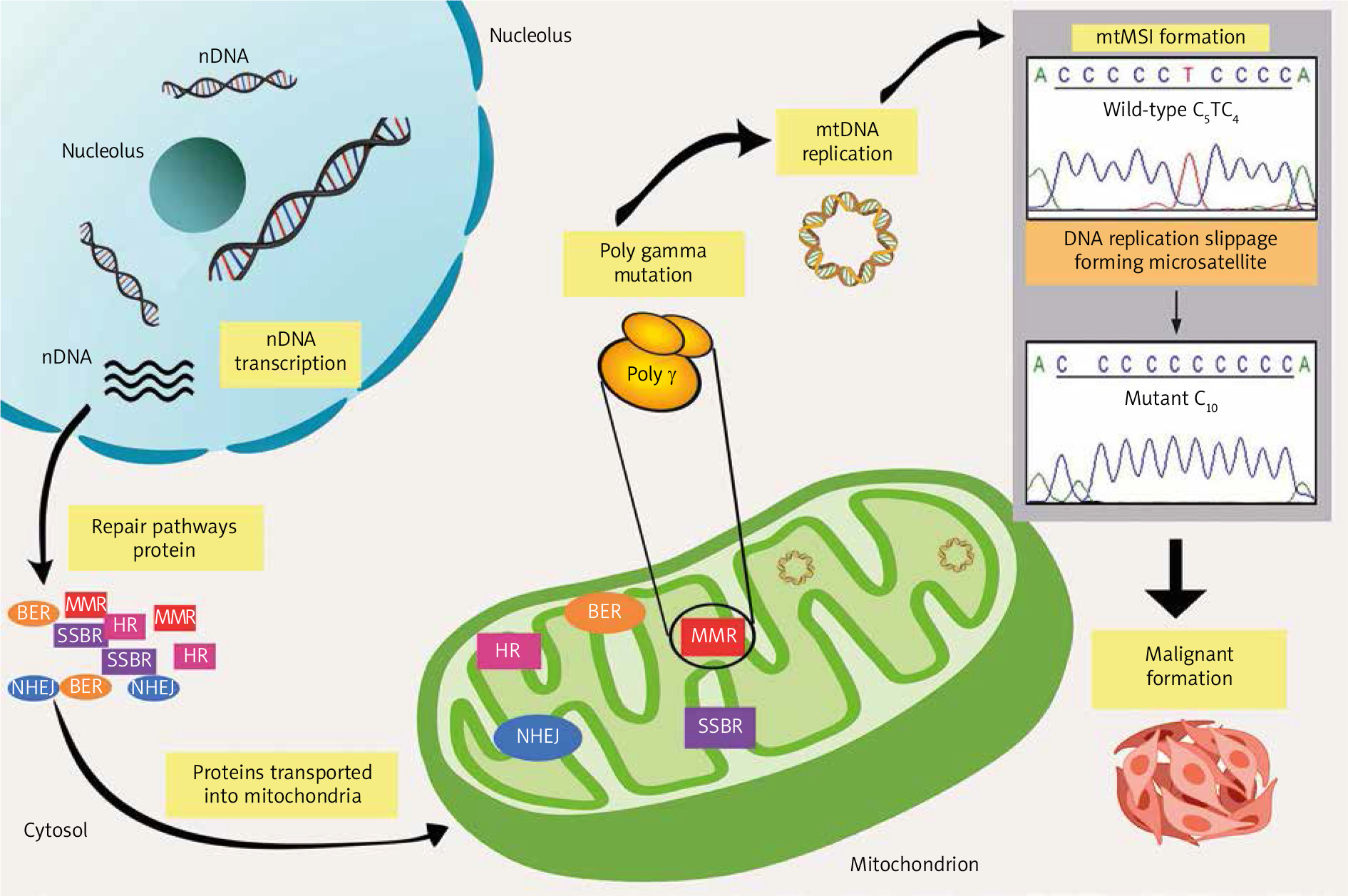
Notably, reliable cellular DNA repair machinery is essential to preserve genomic integrity; thus, the impairment of these structures might affect genome stability and survival. Of note, various DNA repair machinery has been extensively studied in the nucleus. Nevertheless, it has become more evident that mitochondria also experience DNA repair activities, because it has been identified that this organelle occupies almost all the repair mechanisms responsible for the nuclear genome, including break excision repair, single-strand break repair, MMR, non-homologous end-joining, and possibly homologous recombinant [52] (Fig. 1). In general, the involvement of the mtMSI in neoplasm expansion is a result of the discovery of the erroneous replication that occurred sporadically. During the replication process, mtDNA poly γ is prone to slippage, which further causes constant errors. These errors could increase the levels of mtDNA mutational load if they are not fixed immediately. Therefore, the inefficiency of the repair system might lead to the main contributor to the generation of MSI, which is implicated in malignant transformation and development. Nonetheless, all key elements of mitochondrial repair mechanisms and maintenance rely fully on nuclear-encoded proteins. However, numerous mutations, deletions, and other damage may develop if these repair mechanisms are left impaired and the injuries remain unfixed.
Fig. 1
Overview of pathways for DNA repair in mitochondrial microsatellite instability. Mitochondrial DNA genome maintenance and repair pathways completely rely on the importation of the nuclear-encoded protein. However, the emergence of mitochondrial microsatellite instability formation caused by DNA polymerase slippage expands the errors, which leads to a deficiency in repair pathways. The unfixed errors may increase the mutation rates, which affects mitochondrial biogenesis and finally promotes malignant transformation
The mismatch repair system plays a critical role in recognizing and amending errors of base-base mismatches and slippage mistakes made by polymerases during DNA replication [53]. Mismatch repair includes the detection of the mismatch and targeting the newly synthesized strand for repair by a mechanism of excision and DNA resynthesis, followed by ligation to finalize the process [53]. The existence of MMR activity in mammalian mitochondria was originally observed by Mason et al., who showed that rat liver mitochondrial lysates are capable of amending G-T and G-G mismatches through M13-based assay, revealing significant MMR activity [54]. However, this repair mechanism is independent of a key nuclear factor, MSH2, and the addition of MSH3, MSH6, or MLH1 is also absent in human mitochondria. On the other hand, de Souza-Pinto et al. confirmed that repair factor Y-box binding protein (YB-1), which is significant in nearly all DNA- and mRNA-dependent processes, is also involved in mitochondrial MMR [55]. Factor Y-box binding protein localizes to mitochondria and is responsible for mismatch-binding and mismatch-repair activity [56]. Moreover, defects in YB-1 protein might induce mtDNA mutations, and so the presence of YB-1 protein is critical in maintaining mtDNA stability [56].
Conversely, it was later confirmed that MLH1 also localizes to mitochondria and plays a significant role in mitochondrial biogenesis. It was reported that a malfunction in MLH1 with the inhibition of polγ causes an increase of mtDNA lesions [57]. Furthermore, it was found that the overexpression of MLH1 in retinal endothelial cells elevates the non-coding sequence variants, together with a low rate of respiration and increase in apoptosis, but not through the overexpression of MSH2. Therefore, it was suggested that MLH1 provides a defensive role in mtDNA, while overexpressed MSH2 did not render such benefits to mtDNA lesions [58]. This result is in line with a previous report that the silencing of MLH1 through DNA methylation generates mitochondrial damage [59]. It has been shown that deficiency of MLH1 triggers the disrupted mtDNA metabolism and exhibits a significant reduction in respiratory chain complex I activity subsequent to reduced anti-oxidant response and increased sensitivity to ROS-inducing drugs [60]. These data provide evidence that MLH1 is significantly associated with mtDNA function and maintenance. Taken together, these observations imply that not all the repair elements in nDNA are implemented in mtDNA repair machinery; thus, the repair mechanisms in both nuclear and mitochondrial genomes are distinct from one another.
It has been acknowledged that the failure of mtDNA repair, oxidative insults, and replication errors are the main sources of mitochondrial alteration [61]. Nonetheless, the presence of MSI has been considered as a mutator phenotype, which reflects deficiencies in MMR that potentially increase mutation rates. Until now, the identification of faulty MMR genes has been correlated with nuclear MSI (nMSI), although little evidence is available of the precise mechanisms that may commit to mtMSI formation. However, one of the contributing elements of mtMSI accumulation is the failure of mtDNA polγ.
Mitochondrial DNA genome polγ is the sole DNA polymerase that carries out the repair mechanism and replication of mtDNA with proofreading exonuclease activity [62]. However, mtDNA polγ has low frameshift fidelity and is prone to slippage, thus resulting in lower proofreading efficiency [63]. An early report proposed that malfunction of mtDNA polγ induced by persistent oxidative damage might extend replication errors [62]. As noted previously, mtDNA polγ has become a significant target of oxidative insults, suggesting that oxidation of polγ might decrease the repair capacities and mtDNA replication process [8].
In particular, the exonuclease activity of mtDNA polγ is important to sustain high fidelity during DNA replication [64]. However, several reports have suggested that deficiency of the exonuclease site might disrupt the replication process and lead to mtDNA mutation [65, 66]. In 2000, Zhang et al. revealed that transgenic mice carrying a high-level expression of proofreading-deficient polγ led to the accumulation of mtDNA mutations in cardiac tissue [65]. Additionally, homozygous knock-in mice with expressed proofreading-deficient polγ showed mtDNA mutator phenotype and increased mtDNA mutations correlated with premature aging [66]. Moreover, a more recent study examined the role of oxidative modification on replication fidelity of mtDNA polγ, and it was shown that the exonuclease site of polγ is vulnerable to oxidative insults, which leads to lower expression of mtDNA polγ exonuclease activity and increases the mutation rate. In this regard, the exonuclease active site of polγ is a primary target for oxidation; thus, errors in replication appear to be the main cause of mtDNA changes [61].
Also, mtDNA polγ tends to incorrectly duplicate the homopolymorphic repeats [53]. It has been hypothesized that error rates of mtDNA polγ increase with the increase of homopolymorphic sequence length [63]. Previously, it was revealed that most of the instabilities are in the polymorphic length spectrum, which contributes to the functional loss of mitochondria [30]. Indeed, it is conceivable that reduced replication capacity leads to mismatches and mtDNA mutation, which may cripple mitochondrial function and biogenesis. Thus, a proper MMR system is crucial for mtDNA errors because the erroneousness of this machinery might elevate the rate of mutation [47].
Nonetheless, the emergence of the MSI variant may be caused by the inefficiency of the mitochondrial OXPHOS system which is in charge of the release of ROS. Because the location of the mitochondrial genome is close to the ROS production site and has no shield protein, this organelle is susceptible to mutation. Defects in mtDNA respiration may boost ROS production and further risen in glucose-dependent [67]. This phenomenon might extend the risk of mtDNA mutation and potentially promotes mitochondrial dysfunction. Therefore, it was suggested that mitochondrial alterations are linked with mitochondrial dysfunction leading to neoplasm expansion and progression [67].
It was generally accepted that the defect in mtDNA repair machinery may lead to mitochondrial dysfunction and facilitate tumour progression. However, since all repair elements in mtDNA are imported by nuclear-encoded protein, failure in mitochondria may also affect the stability of nDNA. Therefore, several attempts have been made to assess the related mechanisms that drive the formation of nMSI and mtMSI [30, 68–72]. The first study was done by Habano et al., which observed a strong correlation of repair mechanism in mtMSI and nMSI in gastric carcinoma [68]. Since then a few studies have been carried out focusing on the potential correlation of nuclear and mitochondrial genome instability; however, they failed to document the associations. Based on an analysis by Richard et al. in 2000, the replication of the nuclear and mitochondrial genome in cancer cells causes the existence of slipped strand mispairing in breast cancer [69]. However, they found no correlation between mitochondrial and nuclear instability, suggesting that distinct mechanisms are responsible for these instabilities in the development of human breast cancer [69].
Similarly, a previous report also determined that instability in D310 was not related to nMSI, implying a different driver of the incidence [70]. This result corroborates the fact that nMSI has been associated with the dysfunction of MMR genes, and a similar defect may be the factor for mtMSI formation. However, the precise MMR mechanism responsible for mtDNA remains elusive [70]. Meanwhile, a recent study has presented that nMSI and mtMSI are separate events, suggesting that enzymes of MMR are not active in the amendment of mtDNA base mispairing [30]. The authors also stated that nMMR proteins do not have any mitochondrial localization signal and are not detectable by immunohistochemical analysis. This sequence of events implies that there are specific mechanisms for the formation and repair of MSI in the nuclear and mitochondrial genome, respectively. Several elements found in the nuclear genome are not entirely engaged in the mitochondrial MMR system. However, additional evidence of the specific roles of genes and pathways involved in mitochondrial instability is warranted because it would enhance the detailed understanding of mitochondrial repair connected with tumourigenesis.
Mitochondrial microsatellite instability hotspots
The majority of the mtMSI studies have extensively focused on the 3 mtDNA microsatellite markers, such as 303 poly-C (CCCCCCCTCCCCC from position 303–315, 7CT5C), 514 (CA) repeat (CACACACACA from position 514–523, (CA)5 repeats), and 16189 poly-C (CCCCCTCCCC from position 16184–16193, 5CT4C) in the D-loop region.
The poly-C repeat located between 303 and 315 nucleotides and interrupted by a T base at position 310 (termed as D310) has been recognized as a frequent hotspot of deletion/insertion mutations in various multifactorial diseases and cancers [73–78]. D310 alterations were initially discovered in 2001 by Sanchez-Cespedes et al. in several types of tumours [75].
The D310 is crucial for the replication primer binding site and a part of the ‘conserved sequence block II’ (CBS II) of the hypervariable region II (HVII), which is involved in the formation of a persistent RNA-DNA hybrid that results in the initiation of mtDNA heavy-strand replication [79]. Alterations in the mtDNA replication rate can result from the deletion or insertion in the number of the C residues in the poly-C repeat, which may interfere with the binding of mtDNA polymerase γ and other trans-acting elements. It is believed that D310 plays a vital role in preserving the number of mtDNA content.
Moreover, the proper function of mtDNA transcription requires a precise CBS II sequence [80]. When instabilities occur in the mtDNA sequences between 282–300 or 304–300 nucleotides, these can lead to premature transcriptional termination or reduced transcription. Additionally, complete transcriptional termination appears in the alterations of 289–319 nucleotides. Consequently, sequence variations in this repeat, if not repaired correctly, could lead to the disablement of mitochondrial function, increased ROS generation, and eventually may promote tumourigenesis.
In 2003, Mambo et al. determined that the D310 region is responsive to damage when treated with the oxidant agents 4-nitroquinoline 1-oxide and tert-butyl hydroperoxide [81]. They claimed that the occurrence of D310 alterations could be due to the combination of hypersensitivity to oxidative stress and mtDNA repair mechanism ineffectiveness. These discoveries may elucidate the high frequency of homoplasmic D310 somatic mutations identified in different kinds of cancers.
Another poly-C stretch that is also known as mtMSI and mutational hotspot region is the D16189 poly-C tract of the hypervariable region I (HVI), which is situated between 16184 and 16193 nucleotides and is interrupted by a thymine residue at position 16189. Substitution of T to C at 16189 (T16189C) frequently generates a continuous homopolymeric tract of 10 cytosines. This poly-C length instability is often appeared in a heteroplasmic form [37].
Several studies have reported the correlation between D16189 polymorphism and various multifactorial diseases such as metabolic syndrome [82], type 2 diabetes mellitus [82, 83], coronary artery disease [84], and cancers [85, 86]. In a gynaecological cancer study, Liu et al. showed that endometrial cancer patients had a significantly higher D16189 variant frequency (43.1%) compared to non-endometrial cancer [85]. In 2014, Tipirisetti et al. investigated the entire mtDNA D-loop region in the South Indian population with breast cancer and determined that the D16189 variant frequency was significantly elevated in cases compared to controls [86].
D16189 polymorphism has also been claimed to be associated with mtDNA copy number alterations [87]. Based on a study of 837 healthy adult subjects, Liou et al. demonstrated that the mtDNA copy number was reduced in individuals harbouring a variant of uninterrupted poly-C [88]. However, elevated mtDNA content was determined in individuals with diverse interrupted poly-C compared to wild-type individuals [88].
Due to their location in the 7S DNA binding site, D16189 loci appear to play an essential role in mtDNA synthesis regulation. It is also believed that the interrupted poly-C tract seems more effective in attracting and binding to the mitochondrial single-strand binding protein (mtSSB) in comparison to the uninterrupted variant. Therefore, the uninterrupted variant could cause a reduction in the mtDNA replication efficiency with an associated depletion in the quantity of mtDNA. Depleted mtDNA content could affect OXPHOS capacity and further increase ROS production, thereby contributing to some multifactorial disorders [86, 88].
D514 is another MSI hotspot region of the mtDNA D-loop, which is located within 5 CA dinucleotide repeats between nucleotide positions 514 and 523 in the hypervariable region III (HVIII). Researchers report that the D514 genomic instability rate varies in common solid cancer types such as breast [69], gastric [89], glioblastoma [90], ovarian [91], cervical [92], thyroid [93], and nasopharyngeal [94]. It is postulated that CA dinucleotide repeat length alterations may influence mtDNA transcription efficiency due to mtDNA D-loop containing the LSP and 2 HSPs (HSP1 and HSP2). The D514 polymorphism has been suggested to be associated with breast cancer risk and poor survival [95].
The D310, D16189, and D514 are well-recognized as MSI markers in the mtDNA region and have been implicated in the development of many cancers; however, their exact functional mechanism is still unclear. Importantly, additional research is needed to address these unresolved matters about the functional mechanism of mtMSI markers in tumourigenesis.
Mitochondrial microsatellite instability frequencies in solid cancer
In 1998, Habano et al. [47], pioneers in the research of mtMSI in cancer, mainly in colorectal cancer, demonstrated that more attention must be directed toward the role of the mitochondrial genome in carcinogenesis. Since then, many studies have been conducted, determining the link between mtMSI and various types of cancer (Table 1).
Table 1
Summary of studies in which mitochondrial microsatellite instability was examined in some types of solid cancers
| Cancer type | Case number | mtMSI frequency | Country | Author (refs.) | |
|---|---|---|---|---|---|
| D-loop | Other/coding region | ||||
| Colorectal cancer | 45 | 20/45 = 44.4% | 3/45 = 6.7% (ND1 and ND5) | Japan | Habano et al., 1998 [47] |
| 45 | – | 7/45 = 15.6% (ND1 and ND5) | Japan | Habano et al., 1999 [96] | |
| 25 | 7/25 = 28% (D310) | – | USA | Sanchez-Cespedes et al., 2001 [75] | |
| 95 | 32/95 = 33.6% (D310) | – | Norway | Guleng et al., 2005 [70] | |
| 138 | 11/133 = 8.3% (D310) | – | Japan | Kose et al., 2005 [98] | |
| 25 | 9/25 = 36% | – | Taiwan | Lee et al., 2005 [99] | |
| 11 | 6/11 = 54.5% (D310) | – | France | Lièvre et al., 2005 [104] | |
| 365 | 132/365 = 36.2% (D310) 16/178 = 9% (C7TC6) 56/113 = 49.6% (C8TC6) 11/15 = 73.3% (C9TC6) | France | Lièvre et al., 2005 [104] | ||
| 48 | Epithelia 13/48 = 27.1% Stroma 5/48 = 10.4% | – | Korea | Kim et al., 2006 [105] | |
| 62 | 17/62 = 27.4% (D310) | – | France | Legras et al., 2008 [107] | |
| 194 | 47/194 = 24.2% (D310) | – | Taiwan | Chang et al., 2009 [108] | |
| 38 RC 25 SC | 34.3% (D310) 37.5% (D310) | 2.6% (ND1), 5.3% (ND5) 8% (ND5) | Portugal | Pinheiro et al., 2009 [106] | |
| 83 | 23/83 = 27.7% | – | Taiwan | Tsai et al., 2009 [103] | |
| 54 | 25/54 = 46.3% 19/54 = 35.2% (D310) 10/54 = 18.5% (D16184) 4/54 = 7.4% (D514) | – | Korea | Lim et al., 2012 [101] | |
| 44 | 19/44 = 43.2% (D310) | – | Vietnamese | Miwata et al., 2014 [100] | |
| 133 | 11/133 = 8.3% (D310) | – | Japanese | Miwata et al., 2014 [100] | |
| 30/100 = 30% | – | Korea | Lee et al., 2015 [102] | ||
| 182 | 99/182 = 54.4% 92/182 = 50.5% (D310) 9/182 = 4.9% (D514) 3/182 = 1.6% (D16184) | 2/182 = 1.1% (ND1) 7/153 = 4.6% (ND5) | Netherlands | Venderbosch et al., 2015 [30] | |
| 120 | 45/120 = 37.5% 35/120 = 29.2% (D310) 4/120 = 3.3% (D514) 9/120 = 7.5% (D16184) | – | Norway | Kleist et al., 2017 [78] | |
| 21 CC 30 RC | 68.6% (D310) 13.8% (D16184) 47% (D310) 20.9% (D16184) | – | China | Wang et al., 2018 [97] | |
| Gastric cancer | 62 | 10/62 = 16.1% | – | Japan | Habano et al., 2000 [68] |
| 32 | 6/32 = 18.8% | – | Portugal | Máximo et al., 2001 [89] | |
| 8 | 5/8 = 62.5% (D310) | – | USA | Sanchez-Cespedes et al., 2001 [75] | |
| 22 | 13/22 = 59.1% | – | China | Han et al., 2003 [109] | |
| 30 | 11/30 = 36.7% | – | China | Ling et al., 2003 [110] | |
| 68 | 26/68 = 38.2% | – | China | Ling et al., 2004 [111] | |
| 94 | 14/94 = 15% (D310) | – | Japan | Kose et al., 2005 [98] | |
| 31 | 10/31 = 32.3% | – | Taiwan | Lee et al., 2005 [99] | |
| 20 | 4 mtMSI in 18 mutations (7/20 = 35% mutation rate) | – | China | Zhao et al., 2005 [112] | |
| 62 | 10/62 = 16.1% (D310) | – | Japan | Kose et al., 2006 [113] | |
| 100 | 11/100 = 11% | – | Italy | Gargano et al., 2007 [114] | |
| 49 | 4/49 = 8.2% (5/49 = 10.2%, overall mtMSI) | 1/49 = 2% (ND2) | Korea | Jeong et al., 2010 [115] | |
| 122 | 28/122 = 23% | – | China | Ling et al., 2016 [116] | |
| 18 | 40.3% (D310) 37.2% (D514) 11.7% (D16184) | – | China | Wang et al., 2018 [97] | |
| Breast cancer | 40 | 17/40 = 42.5% | – | Argentina | Richard et al., 2000 [69] |
| 17 | 5/17 = 29.4% (D310) | – | USA | Sanchez-Cespedes et al., 2001 [75] | |
| 64 | 12/64 = 19% (D310) | – | Italy & USA | Parrella et al., 2001 [117] | |
| 20 | 3/20 = 15% (D310) | – | Italy | Parrella et al., 2003 [118] | |
| 51 | 15/51 = 29.4% | – | Hong Kong | Wang et al., 2006 [120] | |
| 60 | 13/60 = 21.7% (D310) | – | Taiwan | Tseng et al., 2006 [121] | |
| 64 | 17/64 = 26.6% (D310) | – | Brazil | Santos et al., 2012 [122] | |
| Oesophageal cancer | 38 | 11/38 = 28.9% (D310) (13/38 = 34.2%, somatic mutation) | – | Japan | Kumimoto et al., 2004 [76] |
| 82 | 7/51 = 14% (D310) | – | Japan | Kose et al., 2005 [98] | |
| 72 | 56/72 = 77.8% (D310) | – | Taiwan | Lin et al., 2010 [125] | |
| 66 | 44/66 = 66.7% (D310) | – | Taiwan | Lin et al., 2012 [126] | |
| Hepatocellular carcinoma | 19 | 8/19 = 42.1% (D310) | – | Japan | Nomoto et al., 2002 [136] |
| 52 (11/52=21.2%, overall mtMSI) | 10/52 = 19.2% | 5/52 = 9.6% | China | Fang et al., 2004 [137] | |
| 54 | 10/54 = 18.5% | – | Taiwan | Lee et al., 2005 [99] | |
| Gynaecological cancer | 15 ovarian | 3/15 = 20% | – | Hong Kong | Liu et al., 2001 [91] |
| 50 endometrial (25/50=50%, overall mtMSI) | 16/50 = 32% (D310) 1/50 = 2% (D514) 7/50 = 14% (D16184) | 4/50 = 8% (12S rRNA) | Hong Kong | Liu et al., 2003 [85] | |
| 14 cervical 6 endometrial | 5/14 = 35.7% (D310) 1/6 = 16.7% (D310) | – | Italy | Parrella et al., 2003 [118] | |
| 71 cervical 62 endometrial 78 ovarian | 17/71 = 23.9% (D310) 2/71 = 2.8% (D514) 20/62 = 32.3% (D310) 1/62 = 1.6% (D514) 9/62 = 14.5% (D16184) 14/78 = 17.9% (D310) 5/78 = 6.4% (D514) | 4/62 = 6.5% (12S rRNA) | Hong Kong | Wang et al., 2005 [80] | |
| 24 cervical (9/24=37.5%, overall mutations) | 8 mtMSI in 6 patients 6/24 = 25% | – | China | Chen and Zhan, 2009 [138] | |
| 40 cervical (12/40=30%, overall mutations) | 4 mtMSI in overall mutations | – | China | Zhao et al., 2010 [139] | |
| Lung cancer | 100 | 16/100 = 16% (D310) | – | USA | Sanchez-Cespedes et al., 2001 [75] |
| 31 | 4/31 = 13% | – | Taiwan | Lee et al., 2005 [99] | |
| 37 (12/37=32.4%, overall mtMSI) | 7/37 = 18.9% (D310) 1/37 = 2.7% (D514) 5/37 = 13.5% (D16184) | – | China | Dai et al., 2013 [145] | |
| 48 ADC 42 SCC | 10/48 = 20.8% 8/42 = 19% | – | Korea | Lee et al., 2015 [71] | |
| Thyroid cancer | 126 rTT 40 sTT | 2/126 = 1.6% (D310) 6/40 = 15% (D310) | – | Belarus Germany | Lohrer et al., 2002 [132] |
| 72 | 5/72 = 6.9% (D310) | – | USA | Tong et al., 2003 [133] | |
| 66 (32/66=48.5%, overall mtMSI) | 20/66 = 30.3% (D310) 15/66 = 22.7% (D514) 3/66 = 4.6% (D568) | – | Portugal | Máximo et al., 2005 [93] | |
| Thyroid cancer | 77 | 8/77 = 10.4% (D310) | – | China | Ding et al., 2010 [134] |
| 251 (108 HTNs, 95 CTNs, 48 PTCs) | 39/251 = 15.5% (D310) 17/251 = 6.8% (D514) 4/251 = 1.6% (D568) 11/251 = 4.4% (D16184) | – | Turkey | Bircan et al., 2019 [135] | |
| Head and neck squamous cell cancer | 51 | 19/51 = 37.3% (D310) | – | USA | Sanchez-Cespedes et al., 2001 [75] |
| 93 | 34/93 = 36.6% (D310) | – | USA | Ha et al., 2002 [127] | |
| 18 | 8/18 = 44.4% (D310) | – | Taiwan | Tan et al., 2003 [128] | |
| 109 | 19/109 = 17.4% (D310) | – | France | Lièvre et al., 2006 [130] | |
| 8 | 2/8 = 25% (D310) | – | China | Shu et al., 2019 [131] | |
| Brain cancer | 55 (50 GBM, 4 AII, 1 AIII) | 5/55 = 9% | – | Germany | Kirches et al, 2001 [90] |
| 17 GBM (6/17 = 35.3%, overall mtMSI) | 2/17 = 11.8% (D310) 1/17 = 5.9% (D514) 4/17 = 23.5% (D16184) | – | Germany | Kirches et al., 2001 [90] | |
| 40 (8/40 = 20%, overall mtMSI) | 6/40 = 15% (D310) 2/40 = 5% (D16184) | – | Malaysia | Radzak et al., 2021 [141] | |
| Prostate cancer | 16 | 8/16 = 50% (D310) | – | USA | Chen et al., 2002 [142] |
| Gall bladder | 123 | 47/123 = 38.2% (D310) | – | Chile | Tang et al., 2004 [146] |
| Urinary bladder and renal cancer | 31 BC 21 RLC | 4/31 = 12.9% (D310) 3/21 = 14.3% (D310) | – | Japan | Wada et al., 2006 [147] |
| 16 BTCC | 4/16 = 25% (D310) | – | Italy | Parrella et al., 2003 [118] | |
| Skin cancer | 57 KA 43 SCC | 2/57 = 3.5% (D514) 8/43 = 18.6% (D310) | – | Korea | Alam et al., 2021 [148] |
[i] AII – astrocytoma WHO grade II, AIII – astrocytoma WHO grade III, ADC – adenocarcinoma, BC – bladder cancer, BTCC – bladder transitional cell carcinoma, CC – colon cancer, CTNs – cold thyroid nodules, D310 – (poly-C)n, between np 303-315, D514 – (CA)n, between np 514-523, D568 – (poly-C) n, between np 568-573, D16184 – (poly-C)n, between np 16184-1619, GBM – glioblastomas, HTNs – hot thyroid nodules, KA – keratoacanthoma, mtMSI – mitochondrial microsatellite instability, PTCs – papillary thyroid carcinomas, RC – rectal carcinoma, RLC – renal cancer, rTT – radiation-associated thyroid tumour, SC – sigmoid carcinoma, SCC – squamous cell carcinoma, sTT – sporadic thyroid tumour
Colorectal cancer
Globally, colorectal cancer has been ranked in third place among the commonest malignancies, and based on the World Health Organization GLOBOCAN database there are over 1.84 million predicted with new cases and around 880,792 deaths in 2018 (https://gco.iarc.fr/today/). Mitochondrial MSI is commonly studied in colorectal cancer, and about 8–69% of patients with colorectal cancer have mtMSI. The first study on the involvement of MSI in the mitochondrial genome of colorectal carcinomas was reported by Habano et al., who screened 9 mtMSI sites in 45 sporadic colorectal carcinomas and revealed a mtMSI frequency of 44% (20/45) [47]. They also identified 3 cases with frameshift mutations that corresponded to polyadenosine (A)8 or polycytidine (C)6 tract in the mitochondrial NADH dehydrogenase (ND) genes (ND1 and ND5), which were predicted to result in truncated of complex I proteins. One year later, the same group extended the study by analysing the entire mtDNA amplification scanning in the same series of 45 carcinomas [96]. They found ND1 or N5 gene alterations in 15.6% (7/45) of cases, which included 3 frameshift mutations in the mononucleotide repeat sequences, 2 missense mutations, and one small 15-bp deletion. Six of 7 cases with mutations also presented mtMSI of the (C)n sequence in the D-loop region [96].
Studies by Venderbosch et al. [30] and Wang et al. [97] have shown a high frequency of mtMSI, at 54.4% and 82.4%, respectively. However, in contrast to these, in a study by Kose et al. only 8.3% (11/133) of mtMSI was identified in a large panel of 138 colorectal cancer samples [98].
The frequency of mtMSI in colorectal cancers also varies from community to community. Mitochondrial MSI frequency is 36%, 8.3%, and 43.2% in Taiwanese [99], Japanese, and Vietnamese patients [100], respectively. Lim et al. also reported a high frequency of mtMSI 46.3% (25/54) in Korean colorectal cancer, which mainly occurred in D310 (35%) and D16184 poly C (19%) [101]. In another study, Lee et al. in 2015 [102] reported a 30% frequency of mtMSI in the Korean group of colorectal cancer, which was similar to that found by Tsai et al. [103].
Also recognized as a mutational hotspot in some tumour types, mtMSI in the D310 site is commonly found in several types of colorectal carcinomas. In the year 2001, Sanchez-Cespedes et al. were the first to uncover D310 as a mutational hotspot in several kinds of primary tumours [75]. A study by Sanchez-Cespedes et al. that analysed 25 colorectal tumours showed that 28% of tumours harboured D310 mtMSI.
In 2005, Lièvre et al. conducted an experiment to investigate the presence of D-loop mutations in a series of 365 colorectal cancer patients and found that 36.2% of the cases carried alterations in the D310 microsatellite sequence [104]. They also demonstrated that the prevalence of D310 alterations significantly increased with the number of cytosines in the sequence, in which C7TC6 was 9% (16/178), C8TC6 was 49.6% (56/113), and C9TC6 was 73.3% (11/15), respectively. While, in the same year, D310 alterations together with repeats in the ND1 and ND5 genes were examined by Guleng et al. in a group of 95 colorectal cancer patients and 95 control subjects [70]. The authors detected that 34% of the carcinomas had D310 alterations, while there were only 2% in the control group. In addition, they failed to detect any mtMSI in the ND1 and ND5 genes and concluded that D310 instability was not correlated with nMSI in their cases [70]. In another study, also in the same year, Lee et al. analysed the nucleotide sequence of the D-loop as well as mtDNA content in a series of solid tumours that comprised of 25 colorectal cancers [99]. Impressively, the results indicated that 40% (10/25) of the cases studied had mutations in the D-loop with a 90% (9/10) rate of altered D310 microsatellite sequence. They proposed that mtDNA instability, in conjunction with the diminish in the mtDNA content, plays a crucial role in human carcinogenesis [99].
In 2006, Kim et al. used PCR-SSCP and DNA sequencing analysis to examine 48 micro-dissected cancer epithelia and adjacent stroma of sporadic colorectal cancer [105]. They identified that 27.1% (13/48) of cancer epithelia and 10.4% (5/48) of stroma contained D-loop mtMSI. Three years later, Pinheiro et al. aimed to analyse D310 as well as ND1 and ND5 for MSI in a series of 38 rectal carcinomas in comparison with 25 sigmoid carcinomas [106]. They revealed a D310 MSI frequency of 34.3% and 37.5% of rectal and sigmoid carcinomas, respectively, while ND5 alterations were observed in 5.3% and 8% of rectal and sigmoid carcinomas, respectively. Moreover, only rectal carcinoma cases were reported to harbour ND1 alterations at an even lower frequency, with a MSI frequency of 2.6%.
The prevalence of D310 alterations in colorectal adenomas and liver metastases of colorectal cancers were reported in 27.4% (17/62) and 33.3% (5/15), respectively, in a study performed by Legras et al. [107]. Their investigations were continued with a case-control study by assessing the possible role of D310 polymorphism in colorectal adenoma risk. The authors proved that germline D310 polymorphism might not be a risk factor for colorectal adenomas. Nevertheless, they suggested that D310 mutation could be considered as a potential biomarker for the early detection of colorectal cancer.
Several studies have evaluated the correlation of mtMSI with clinicopathological parameters of colorectal cancer. Lee et al. found mtMSI in 30% of 100 colorectal cancers and later demonstrated that patients with mtMSI had shorter overall survival [102].
In a 2017 study carried out by Kleist et al. detected mtMSI in 37.5%, and this was the first to show that D-loop MSI was significantly associated with TP53 wild-type status of primary colorectal cancer [78]. Additionally, D-loop MSI was not associated with gender, clinical stage, tumour location, survival or KRAS, NRAS, BRAF, or PIK3CA mutations.
Several studies have investigated the prognostic value of mtMSI in colorectal cancer; however, their results were considered controversial. In a previous study, Chang et al. reported that mtMSI had no prognostic effect in colorectal cancer patients [108]. In contrast, a study by Tsai et al. revealed that Dukes’ stage C colorectal cancer patients with mtMSI had better disease-free and overall survival [103]. They concluded that mtMSI provides a good prognosis in Dukes’ C colorectal cancer. In a study by Lim et al., it was shown that there was a higher frequency of mtMSI in larger size tumours and more advanced tumor node metastasis stage, indicating mtMSI as a risk factor for unfavourable outcome [101].
Gastric cancer
The prevalence of mtMSI in gastric cancers is approximately 10-63%. An initial report of mtMSIs in gastric cancer was demonstrated by Habano et al. in 2000 [68]. In their study, 10 out of 62 (16.1%) Japanese gastric carcinoma cases had the mtMSI phenotype in the D-loop region. They believed that the mtMSI phenotype exhibited in some gastric carcinoma cases might play a significant role in tumourigenesis.
In the following year, a study performed by Máximo et al. identified mtMSI in 18.8% of 32 gastric carcinomas [89].
In the same year, Sanchez-Cespedes et al. reported a higher frequency of mtMSI (62.5%) in primary gastric tumour patients [75]. This observation was further supported by a report from Han et al. [109]. They analysed 22 patients with gastric tumours in China and discovered that 59.1% of tumours and 40.9% of the adjacent normal tissues carried the D310 or D16184 instabilities.
In the year 2003 Ling et al. observed a 36.7% (11/30) frequency of mtMSI in gastric cancer [110]. They also demonstrated that mtMSI-positive gastric cancer had significantly higher interleukin-8 activity levels than mtMSI-negative gastric cancer. One year later, the same group extended the study by examining 68 cases of advanced gastric cancer, 40 cases of chronic gastritis, 30 cases of intestinal metaplasia, and 20 cases of dysplasia. They found that a progressive accumulation of mtMSI occurred in the histological progression from chronic gastritis to cancer through intestinal metaplasia and dysplasia (12.5−38.2%) [111]. Interestingly, this study also observed a statistically significant association between mtMSI with intestinal-type and distal location of gastric cancer.
In 2005, 3 groups of researchers from Taiwan, Japan, and China examined mtMSI in gastric cancer. Lee et al. reported the frequency of mtMSI to be 32.3% in 31 gastric cancers with alterations in the mononucleotide and dinucleotide repeat of D310 [99]. Kose et al. published their report on using microsatellite assay investigating 96 Japanese patients and identified an mtMSI rate of 15% at the D310 region [98]. Zhoa et al. analysed the D-loop region mutation in 20 Chinese specimens and found sequence mutations in 35% of the specimens including 4 microsatellite instabilities in the mutations [112].
In 2006, Kose’s team expanded the microsatellite instabilities study by examining 74 gastrointestinal stromal tumours for mtMSI at D310 as well as nMSI at BAT26 [113]. The authors noted that the MSI percentage at D310 was 16%, higher than that at BAT26 (5%), despite the lack of statistical significance between them in gastrointestinal stromal tumours.
The first endeavour to explore a possible connection between the RUNX3 promoter methylation, nMSI, and mtMSI in gastric cancer was attempted by Gargano et al. and was published in 2007 [114]. One hundred cases of gastric cancer were selected for the nMSI and mtMSI analysis using the National Cancer Institute guidelines (the Bethesda-recommended panels of microsatellite markers) and D310, respectively. According to their work, mtMSI was identified with a frequency of 11% while high-level nMSI was detected in 13% of cases. This study also highlighted that mtMSI was significantly associated with tumour-node-metastasis staging. Furthermore, an essential correlation between MSI-H status, mtMSI, and RUNX3 methylation was demonstrated, proposing a potential connection between these events in gastric carcinogenesis.
In a series of 49 gastric cancer patients from the general surgery division at Dongsan Medical Centre, Jeong et al. reported a mtMSI rate of 10.2% [115]. Moreover, this study also showed that all mtMSI were significantly observed in intestinal-type cancer. Interestingly, the author also noted a similar frequency of mtMSI occurring in gastric dysplasia as a precancerous lesion (12.5%, 3/24), suggesting that mtMSI might contribute to the early stages in the progression of gastric carcinogenesis.
In the year 2016, Ling et al. explored the association of mtMSI with Helicobacter pylori (H. pylori) infection and the level of interleukin-8 (IL-8) in gastric cancer [116]. They published the results of their analysis regarding mtMSI in 122 samples of gastric carcinomas and found a mtMSI rate of 23%. According to their study, an important significant association was revealed between mtMSI, H. pylori infection, and increased IL-8 level. The authors concluded that mtMSI might play a critical role in different mechanisms of gastric cancer development.
In a recent study, Wang et al. used a sequencing assay to detect mtMSI in the whole mtDNA D-loop in 69 gastrointestinal cancers in Chinese patients, of whom 18 patients had gastric cancer [97]. They reported a rate of mtMSI in the D16184, D514, and D310 regions of 11.7%, 37.2%, and 40.3%, respectively.
Breast cancer
Worldwide, breast cancer is the most diagnosed cancer among women and one of the most significant causes of death among them. mtMSI frequency in breast cancer has ranged from 10.8% to 42.5% in various studies.
The initial study on the occurrence of mtMSI in breast cancer was published in 2000 by Richard et al. [69]. Tumour and matched normal tissues of 40 patients were analysed for the D514 microsatellite site and 4 MnlI sites located between the 16,108- and 16,420-bp positions of the D-loop region. They revealed a MnlI site alterations frequency of 47.5% in the tumours and this rate was 216-fold greater than the spontaneous rate in the female germline. Furthermore, the mtMSI rate was shown to be 42.5%, which indicated a 16-fold higher rate of spontaneous mutations.
Mitochondrial DNA genome D-loop region, particularly at D310, was observed to be a target area that is mostly found altered in breast cancer. In 2001, Parrella et al. performed direct sequencing analysis to investigate the frequency and distribution of mtDNA mutations in 18 primary breast cancers [117]. Eleven tumours were reported to harbour 12 somatic mutations at a mutational frequency of 61%, with 5 alterations in a homopolymeric C-stretch at D310. Moreover, a rapid PCR-based assay revealed 7 more D310 alterations in 46 additional cases of breast tumours, which resulted in a D310 alteration rate of 19% (12/64) in overall breast tumour cases. In 2003, a further study of D310 instability was carried out by Parrella et al. in 4 different tumour types, which included 20 cases of breast tumour [118]. The author later determined a 15% D310 abnormality percentage in breast tumour cases.
In another study, Alazzouzi et al. carried out an extensive experiment to investigate the alternative patterns of microsatellite alterations in cancer cells by analysing mtMSI as well as nMSI in breast ductal adenocarcinomas [119]. They revealed results in which no MSI was identified in any of the dinucleotide and mononucleotide repeats of the nuclear genome in these tumours. Also, the frequency of mtMSI was determined to be 10.8% with the alterations that occurred in the transcription control region. None of the mitochondrial alterations was observed in the normal matched tissues, showing that this mtMSI appeared as sporadic somatic mutations in the tumour cells.
In 2006, Wang’s group screened the stability status of 12 mtDNA microsatellite loci in a series of common female cancers and revealed that 29.4% had a positive instability in breast cancer [120]. Instability was discovered at 3 of 12 microsatellite markers, which were located in the D-loop. According to their data, all mtMSIs in breast cancer were reported at nucleotide positions D310, D514, and D16184, with rates of 13.7%, 7.8%, and 15.7%, respectively. In a different study, Tseng et al. analysed somatic mutations in the D-loop region in 60 Taiwanese women patients by using direct DNA sequencing [121]. They noticed that 30% (18/60) of tumours had somatic mutations, with the majority of alterations (13/18 = 72.2%) being in npD310 poly-C tract.
The genomic instability in both the nuclear and mtDNA of 64 sporadic breast cancer and paired adjacent non-cancerous breast tissues from Brazilian patients were analysed by Santos et al. using nuclear STR loci genotyping and direct sequencing of the HVI and HVII regions [122]. It was reported that 42.2% of cases had somatic mtDNA alterations, with the most prevalent base change being at region D310 poly-C (17/64 = 26.6%). Additionally, about half of cancer cases harboured nMSI, with the majority at D13S790 loci for the 13q31 region. The authors concluded that instabilities at the nuclear 13q31 locus and in mtDNA may play an important role in the development of breast cancer.
There are also a few studies that report a negative finding of this mtMSI in breast cancer patients. In 2013, Tipirisetti et al. published the results of a study in which mtMSI was not uncovered in any of the 11 mtDNA microsatellite loci analysed in breast cancer patients except at D310, which indicated presumably mutational hotspots rather than mtMSI [123]. This finding is consistent with an earlier study by Tan et al., who reported that no MSI were found in all breast tumours and their matched normal tissues [124].
Oesophageal cancer
Worldwide, oesophageal cancer is the sixth leading cause of cancer mortality. Each year ~572,034 new oesophageal cancer cases are diagnosed and > 508,000 patients die of oesophageal cancer. Similarly to hepatocellular cancer, less attention and research has been focused on the mtMSI in patients with oesophageal cancer.
In 2004, Kumimoto et al. sequenced the HVR-I and HVR-II regions of mtDNA from 38 primary oesophageal tumours and matched normal tissues for the somatic mutation rate [76]. They discovered 14 somatic mutations in 13 patients, and a 28.9% (11/38) mtMSI rate was noted at region D310 poly-C stretch. In another study that involved a series of 82 Japanese cancer cases, Kose et al. reported a prevalence of mtMSI in the D310 region of 14% [98].
A previous study involving oesophageal squamous cell carcinomas among Taiwanese patients conducted by Lin et al. determined a high rate (77.8%, 56/72) of somatic D310 alterations [125]. The author also claimed that these variants were significantly correlated with a poorer prognosis. Two years later, the same group extended their research by examining 66 thoracic oesophageal squamous cell carcinomas for mtMSI and nMSI using microsatellite analysis for markers D310 and D17S960 (TP53), respectively [126]. They observed a higher frequency of 66.7% of samples with the D310 alterations. Moreover, only 6.1% of samples harboured nMSI at D17S960.
Head and neck cancer
Head and neck squamous cell carcinomas (HNSCC), which refer to malignancies of the oral cavity, pharynx, and larynx, are the sixth most common cancer by incidence worldwide. Mitochondrial MSI in HNSCC has been studied by several teams. In the case of premalignant head and neck lesions, Ha et al. analysed the mtMSI in 93 patients and uncovered D310 sequence aberrations at a frequency of 36.6% in lesions [127]. Surprisingly, the author noted that the frequency of mtMSI increased according to the disease progression from benign hyperplasia (21.9%), mild dysplasia (33%), moderate dysplasia (36%), and severe dysplasia (50%), to carcinoma in situ (61.5%). Therefore, these aberrations might be used as markers of clonal progression premalignancy to cancer. Also, the authors examined mtMSI at 6 loci in patients that comprise metachronous and synchronous lesions and found that most cases displayed a clonal relationship between these lesions.
In 2003 a study that involved the entire mtDNA mutation screening using temporal temperature gradient gel electrophoresis and followed by sequencing in oral cancers, Tan et al. reported that 44.4% of the cases (8/18) were found to have insertions or deletions in the D310 repeat [128]. In the subsequent year, Poetsch et al. sequenced 2 parts of the D-loop as well as MTND1 and MTND5 genes in 67 primary HNSCC from 56 patients [129]. They similarly demonstrated D310 repeat instability in 42% of the tumours. The nMSI at IGFIIR, hMSH3, hMSH6, and 5 other dinucleotide repeats were also tested on the same tumours for comparison. The frequency of nMSI (36% low nMSI and 13% high nMSI) determined in this series was less than mtMSI, and no correlation existed between them.
In a large series of 109 HNSCC cases from the Laennec Hospital (Paris, France) was conducted by Lièvre et al. in 2006 [130]. This group revealed a rate of 17.4% (19/109) cases harboured alterations in the D310 repeat. In a more recent study, Shu et al. analysed 10 mtDNA microsatellite markers on 8 tongue squamous cell carcinomas, adjacent non-cancerous tissue, and peripheral blood samples from 8 Chinese patients [131]. The authors determined 25% (2/8) of mtMSI, and they were localized in the D310 region.
Thyroid cancer
Thyroid cancer is the most common neoplasm of the endocrine system, and its incidence has been steadily rising worldwide. Several studies have been carried out to explore the occurrence of mtMSI in thyroid cancer. The first manuscript to be reported concerning mtMSI in thyroid cancer was published in 2002 by Lohrer et al. [132]. Two series of tumour tissues from 126 radiation-associated and 40 sporadic thyroid tumour patients were analysed for the instability of the hypervariable region II of the mtDNA. The authors revealed an extremely low frequency of D310 poly-C tract in 2 out of 126 radiation-associated tumours from Belarus and 6 out of 40 tumours from Munich. Nevertheless, the frequencies were found to be associated with the patient’s age.
A lower percentage of the mtMSI in thyroid cancer has also been reported by Tong et al. [133]. This study involved the analysis of D310 region alterations using PCR-based methods in 72 thyroid cancers, which included papillary, medullary, anaplastic, follicular, and insular thyroid carcinoma. The overall rate of D310 instability was shown to be 6.9%.
In 2005, a study of 66 thyroid tumours comprising benign adenomas, follicular, and papillary carcinomas from a Portuguese series by Máximo et al. further revealed a high rate of mtMSI [93]. These mtMSIs were not limited to cancer cases only but were also identified in benign lesions. The authors detected 48.5% (32/66) of thyroid patients contained somatic instability in D-loop microsatellite regions with the most occurring at D310 (30.3%, 20/66), followed by D514 (22.7%, 15/66), and D568 mononucleotide repetitive (4.6%, 3/66).
In a panel of 101 patients with malignant thyroid carcinomas or non-tumour nodular goitres (benign thyroid adenomas) from the Wenzhou area, Ding et al. discovered a mtMSI rate of 10.4% (8/77) in malignant thyroid carcinomas and 10.5% (2/19) in nodular goitres [134]. This study indicates that mtDNA alterations could play a pivotal role during the initial stage of thyroid tumorigenesis.
In 2019, Bircan et al. observed a higher frequency of D310 somatic mutations in the hot thyroid nodules group (22.2%) than in cold thyroid nodules (10.5%) and papillary thyroid carcinoma samples (10.4%) [135]. Also, a higher percentage of the mtMSI was found in D16184 (20.8%) and D514 (25%) of papillary thyroid carcinoma. They concluded that D310 instability might be utilized as a diagnostic clonal expansion biomarker for premalignant thyroid tumour cells and considered D514 as a prognostic biomarker for benign to malign transformation in thyroid tumours [135].
Hepatocellular carcinoma
Hepatocellular carcinoma (HCC) is believed to be the most common primary cancer of the liver and represents the third most common cause of cancer death and the sixth in incidence worldwide. So far, there have been very few studies carried out on the link of mtMSI in hepatocellular carcinogenesis. One of the earliest studies was published in 2002 by Nomoto et al. reported a D310 poly-C tract deletion/insertion alterations frequency of 42.1% (8/19) in Japanese HCC cases [136]. In 2004, and in an effort to report the frequency of mtMSI and nMSI in 52 HCC patients, a study was reported from China by Fang et al. [137]. A rate of 21.2% (11/52) and 5.8% (3/52) of HCC harboured mtMSI and nMSI, respectively. The authors stated that no association was found between both MSIs in the cases of HCC. In a further study that involved a series of solid tumours from Taiwanese patients, Lee et al. published an 18.5% (10/54) mtMSI frequency in HCC cases [99].
Gynaecological cancer
Gynaecological cancer is a major health burden globally for women, and approximately 70,500 women are diagnosed with this cancer each year. The 3 main types of gynaecological cancer are cervical, endometrial, and ovarian. The frequency of mtMSI in gynaecological cancer ranges approximately between 20% and 50%. The mtMSI was first described in ovarian cancer samples (20%, 3/15) by Liu et al. in 2001 [91]. Two years later, a follow-up study of mtMSI was conducted by the authors on 50 samples of primary endometrial carcinomas, revealing a higher rate of mtMSI (50%), particularly detected in 3 different regions (D310, D514, and D16184) of the D-loop region and one in the 12S RNA gene [73].
In the year 2005, Wang et al. investigated the contribution of mtMSI in a series of gynaecological cancers [80]. In this large study that involved 262 pairs of female cancers and matched normal tissues, mtMSI was analysed for 12 microsatellite regions in primary cervical, endometrial, ovarian, and breast carcinomas. Most of the mtMSI belonged to D-loop microsatellite loci (95.6%), particularly in D310, D514, and D16184.
A year later in a follow-up study, Wang et al. assessed mtMSI in another 257 samples of common female cancers, included cervical, endometrial, ovarian, and breast cancers [120]. In agreement with their previous findings, the authors reported the greatest frequency of mtMSI in 48.4% of endometrial cancers followed by 29.4% of breast cancers, 25.4% of cervical cancers, and 21.9% of ovarian cancers.
A 2009 study of 24 cervical carcinomas by Chen and Zhan demonstrated that 9 patients harboured 30 alterations in the D-loop region; 8 (27%) of these changes were mtMSI (8 mtMSI in 6 patients, 25%) [138]. In another study of 40 cervical cancer specimens, Zhao et al. reported that 21 mutations were detected in 12 patients, where there were 4 mtMSI in the mutations [139].
Mitochondrial MSI has also been studied in endometriosis patients by Govatati et al., who claimed that less frequently mtMSI occurred in endometriosis cases [140].
Other cancers
Mitochondrial MSI has also previously been examined by some groups in several other solid cancers such as lung cancer, brain cancer, prostate cancer, bladder cancer, renal cancer, and gallbladder cancer.
Fifty glioma paraffin-embedded tissues were evaluated by Kirches et al. using a combination of laser microdissection and PCR technique for D-loop poly-C tract alteration analysis [90]. The authors determined only 9% of mtMSI frequency in analysed specimens. Also, they conducted direct sequencing of the whole D-loop of 17 frozen glioblastoma samples and matched blood and revealed that 6 of these tumours harboured mtMSI at other repeats in the D-loop region (35.3%). More recently, we detected mtMSI in 20% of a series of 40 primary brain tumours. We observed a total of 8 different alterations within 2 loci, D310 and D16184, of mtMSI markers [141].
Displacement-loop sequence alterations were examined by Chen et al. in 16 patients with prostate cancer [142]. Deletions or insertions appeared in 4 nucleotide positions; 50% of the patient samples showed a mutational hotspot for mtMSI at D310. In 2012, the study of Ashtiani et al. involved sequencing the complete mtDNA D-loop of 40 Iranian prostate cancer cases and benign prostatic hyperplasia patients [143]. They found the insertion of 1 or 2 nucleotide C at D310 was more frequent in both cancer and benign prostatic hyperplasia groups.
Several groups of researchers have addressed mtMSI in lung cancer. The study conducted by Sanchez-Cespedes et al. discovered D310 mtMSI in 16% of 100 lung tumours [75], meanwhile Lee et al. determined in 13% of 31 tumours [99]. In 2003, Suzuki et al. amplified a 336-bp amplicon of the D-loop that contained D310 to detect polymeric C tract instability in 55 non-small cell lung cancers [144]. About 20% (11/55) of the tumours harboured these instabilities. Furthermore, the authors also determined that about 50% of D-loop alterations in 28 lung cancer cell lines occurred in this polymeric C tract. In a previous study, the mtMSI of 37 lung carcinoma and paired adjacent normal lung tissues from Chinese patients were analysed by Dai et al. using PCR-SSCP assay and sequence analysis [145]. It was reported that the percentage of mtMSI in lung carcinoma samples was 32.4% (12/37). Also, the lung carcinoma samples that harboured mtMSI were demonstrated to be correlated with a lower average of mtDNA content compared to cancer tissues without mtMSI. In 2015, an analysis of 48 patients with adenocarcinoma and 42 with squamous cell carcinoma of the lung from Dongsan Medical Centre, Korea uncovered mtMSI at a frequency of 20.8% and 19%, respectively [71].
The occurrence of D310 alterations in gallbladder cancers has been extensively studied by Tang et al. in 2004 [146]. Genetic analysis of archival gallbladder tissues from 123 tumours, 53 dysplasias, 90 histologically normal tissues adjacent to cancer, and 15 normal gallbladders were performed using PCR and sequencing assay for D310 alterations. This study reported a frequency of D310 alterations in the tumour, dysplastic lesions, and normal adjacent tissues of 38.2%, 57.1%, and 45.5%, respectively. Moreover, these D310 insertions/deletions were not only identified in cancer specimens, but the authors also found them in dysplastic and normal epithelial-derived from surrounding chronic cholecystitis (non-tumor) specimens. These outcomes proposed that there might be an early role of D310 alterations in the progressive series pathogenesis of gallbladder cancer.
Wada et al. published the work regarding mtMSI in the urinary bladder and renal cancers in 2006 [147]. They examined genetic alterations in the mtDNA D-loop region as well as oxidative DNA damage by evaluating the level of 8-hydroxy-2’-deoxyguanosine (8-OHdG) in a panel of 31 bladder and 21 renal cancers in Japanese patients. Overall, 12.9% of bladder and 14.3% renal cancer cases had mtMSI, particularly at D310. Importantly, the 8-OHdG content was significantly increased in the cancerous renal tissues as compared with the paired non-cancerous tissues. Nevertheless, many tumour cases with mtMSI harboured lower 8-OHdG content than mtMSI-negative tumours.
A 2021 recently published report by Alam et al. investigated the possible mtMSI in skin cancer, which comprised 57 patients with keratoacanthoma (KA) and 43 patients with squamous cell carcinoma (SCC) [148]. This report determined that mtMSI rates of 3.5% (2/57) of KA patients and 18.6% (8/43) of SCC patients, respectively. The authors also revealed that the mtMSI spot/location was found to be significant with tumour type, whereby the D514 alterations were detected in KA patients, and only SCC patients harboured mtMSI at D310.
Conclusions
Over the past decades, the nDNA has been the focus of studies in the search for cancer susceptibility genes; nevertheless, accumulating evidence now converges to demonstrate that mtDNA may also play an essential role in the genetic aetiology and pathophysiology of cancer. The present review provided a brief overview of several aspects of the mitochondrial genome and then summarized evidence of previous studies concerning the role of mtMSI in most solid cancers.
The study of mtDNA alterations in the context of cancer progression has been recognized for centuries, and there are a vast number of outstanding questions and opportunities in the area that require further investigation. According to cumulative evidence generated from previous studies, this review indicates that mtMSI could contribute to increased cancer risk. However, it is still an ongoing concern whether these instabilities are merely reflecting secondary epiphenomena of the underlying cancer process or comprise a true causative connection. Future larger prospective studies should be conducted to further elucidate the role of mtMSI in solid cancers.






