
Introduction
Type 1 diabetes mellitus (T1DM) is characterised by the immune system’s destruction of insulin-producing β cells, resulting in a reliance on externally administered insulin. The clinical onset, marked by classic hyperglycaemic symptoms, is more commonly observed in adolescents and young individuals, typically when the remaining β-cell population falls below 30%. Treatment for patients primarily involves the administration of exogenous insulin. Although the exact causes of T1DM are not fully understood, the development of this condition is influenced by a combination of genetic and environmental factors. Notably, certain genetic predisposing factors, particularly specific class II haplotypes associated with HLA genes, such as DRB103:01-DQB102:01 (DR3-DQ2) and DRB104:01-DQB103:02 (DR4-DQ8), have been strongly linked to an increased susceptibility to T1DM [1]. These genes are responsible for encoding cell surface proteins involved in immunological functions, specifically antigen presentation, which plays a role in the development of T1DM. Additionally, various environmental factors, including prenatal influences, infections, diet, and stressful events, can contribute to the onset of T1DM, particularly during childhood. Prior to the appearance of symptoms, there is a prolonged asymptomatic period during which the tolerance of the immune system towards β-cells is compromised, leading to the loss of its ability to recognise them as “self”. Subsequently, individuals experience clinical symptoms of T1DM once there is a significant decrease in β-cell mass. In the preclinical phase of the disease, autoantibodies against islet cells can be identified in the bloodstream. Despite the autoimmune response that targets newly formed β-cells, the ability to regenerate β-cells can be maintained for several decades following the clinical onset of T1DM (Fig. 1) [2]. The residual function of β-cells can be evaluated by measuring serum C-peptide levels, which can provide information about the endogenous secretion of insulin even after initiating insulin therapy [3]. In some cases, patients with T1DM may experience a temporary and partial recovery of β-cell function during the course of the disease, which is referred to as the partial clinical recovery phase (PCRP) or commonly known as the “honeymoon period” [4]. The concept of PCRP was initially described by Jackson et al., who observed a rapid decrease in the need for exogenous insulin in diabetic children following regular insulin treatment, often accompanied by episodes of hypoglycaemic shock during this period [2]. During the PCRP, the requirement for external insulin is reduced to less than 50% of the initial levels, and in certain instances, complete independence from insulin therapy can be achieved while maintaining near-normal glycaemic control. However, after the PCRP, individuals with T1DM will still require lifelong insulin treatment, and the likelihood of developing secondary complications remains high in most cases. The occurrence of PCRP is not limited to a specific age group and can be observed in individuals of various ages who exhibit similar symptoms and metabolic characteristics.
Figure 1
Even though there is an autoimmune response that specifically attacks recently developed β-cells, the capacity to regenerate β-cells can persist for many decades after the initial manifestation of T1DM
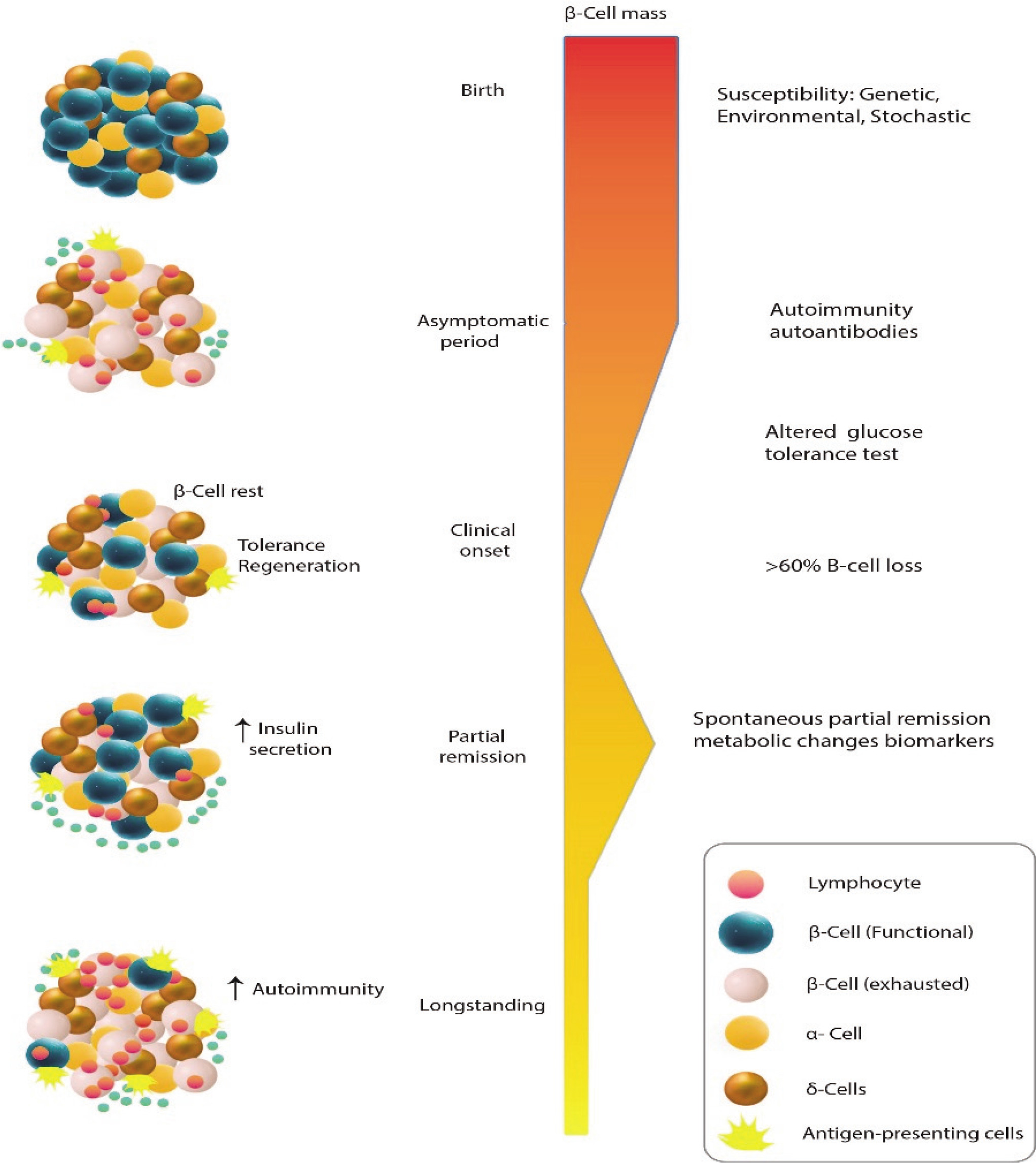
The PCR phase of T1DM exhibits significant heterogeneity, the underlying reasons for which are largely unclear. However, it can be considered a complex sub-trait influenced by a combination of genetic and environmental factors. For instance, a recent study has proposed a potential association between a SNP within prostaglandin receptor EP4 gene (PTGER4) and the remission frequency in T1D patients [5]. The EP4 prostanoid receptor is part of a family of G protein-coupled receptors and is one of the 4 receptor subtypes for prostaglandin E2. Through cAMP effectors such as protein kinase A and exchange protein activated by cAMP, EP4 signalling exerts various physiological and pathophysiological roles. It is widely distributed throughout the body. Although EP4 is known for its anti-inflammatory effects on mononuclear cells and T cells, recent evidence suggests its involvement in proinflammatory actions as well. This review aims to summarise current research findings on the biological functions and role of EP4 receptor in T1DM, with a focus on PCR phase.
Paediatric T1DM: pathophysiology and epidemiology
While T1DM primarily affects children and young individuals [6], it can also develop across various age groups. The timely and attentive management of paediatric T1DM is crucial due to its adverse effects on the well-being of children, their families, and the heightened risk of long-term complications. Paediatric T1DM exhibits distinct characteristics compared to the adult form of the disease. These differences include a reduced residual function of β-cells, elevated levels of anti-insulin antibodies, and a stronger association with HLA types that pose a higher risk [7]. Children who develop T1DM before the age of 7 years face the potential risk of cognitive deficits and impaired mental development. These effects may arise from secondary complications such as episodes of hyperglycaemia and hypoglycaemia, which can have detrimental effects on brain development [8].
T1DM is a medical condition that emerges as a result of the interplay between genetic and environmental factors. The predominant feature of T1DM is the autoimmune deterioration of pancreatic β-cells, which play a crucial role in the production of insulin [9, 10]. Autoimmunity is indicated by the presence of detectable antibodies to ICA512/IA-2, insulin autoantibody (IAA), and glutamic acid decarboxylase (GAD). The progressive destruction of β-cells caused by insulitis leads to pre-diabetes and eventually develops into overt diabetes mellitus. Additionally, individuals with T1DM have an increased susceptibility to developing other autoimmune disorders, such as Hashimoto’s thyroiditis, celiac disease, Addison’s disease, and myasthenia gravis [10]. Extensive genome-wide association studies and meta-analyses have revealed the association of approximately 40 genetic loci with T1DM. Notably, specific genetic loci within the major histocompatibility complex (HLA) region, such as the DR3/4, DQ 0201/0302, DR 4/4, and DQ 0300/0302 alleles, have been identified as having a heightened correlation with an increased susceptibility to T1DM. Estimates suggest that the risk of developing T1DM is approximately 5% when there is a first-degree relative affected by the condition, with a slightly higher risk observed if the affected parent is the father rather than the mother [11]. To date, interventional trials aimed at delaying or preventing the onset of T1DM in genetically at-risk individuals have yielded unsuccessful results. However, international research networks like TrialNet and TRIGR continue to conduct ongoing investigations with the goal of exploring strategies to prevent, delay, or potentially reverse the progression of T1DM [12].
Recent epidemiological evidence concerning paediatric T1DM reveals variations in incidence rates across different regions, with the highest rates observed in Finland and the lowest in East and South-East Asia. Globally, there has been an overall increase in T1DM incidence at a rate of 3–4% per year [13]. Notably, there has been an observed pattern of delayed diagnosis in children between the ages of 0 and 4 years, indicating the potential impact of novel environmental factors that heighten the susceptibility to autoimmune diseases, such as T1DM, within genetically stable populations. This observation aligns with the hygiene hypothesis, which suggests that a lack of early childhood exposure to a diverse range of infections, such as pathogenesis, may hinder the immune system’s maturation, resulting in changes in immunoregulation that increase the risk of T1DM [14]. Additionally, the prenatal environment plays a critical role in sensitising individuals to autoantigens of β-cell and shaping an expression profile of particular gene in CD4+ T cells [15]. Together, these findings indicate that T1DM is a multi-step process that begins early in life. Early identification of T1DM symptoms and timely diagnosis is critical to prevent the occurrence of diabetic ketoacidosis, which is the primary cause of mortality during the initial stages of T1DM. One year after diagnosis, patients diagnosed at a younger age and presenting with ketoacidosis tend to exhibit lower stimulated C-peptide levels, suggesting a decrease in residual β-cell function among these individuals. Moreover, the youngest children have been reported to experience a more rapid loss of β-cell function in comparison to individuals in higher age brackets. This implies that in very young children, there may be either a more aggressive destruction of β-cells or a diminished ability of β-cells to regenerate themselves [16]. Understanding the mechanisms underlying the occurrence or absence of the clinical recovery phase in children with T1DM, referred to as remitters and non-remitters, respectively, is of paramount importance because it significantly influences the long-term prognosis of patients. Notably, one significant challenge in the initial management of T1DM is the lack of a consistent strategy to prevent early dysglycaemia in non-remitters, which includes children and adolescents who do not experience partial clinical remission related to the disease [17–20].
PCR phase: classification, frequency, and mechanisms
The honeymoon period in T1DM can generally be divided into 2 stages: partial clinical recovery (PCRP) and complete clinical recovery (CCRP), both of which are temporary and depend on the extent of β-cell function recovery. Limited epidemiological studies have been conducted on PCRP, and the findings indicate a very low occurrence rate ranging from 0% to 3.2% [21]. On average, the complete recovery period lasts approximately 3 months [22]. In contrast, most studies focusing on the honeymoon period have been conducted on PCRP, revealing occurrence rates of 35% to 43%, although these percentages vary significantly among different countries [17, 23]. Additionally, research has shown that the peak prevalence of PCRP typically occurs 3 to 6 months after initiating insulin therapy, and as the disease progresses, the recovery rate declines [24]. As for the PCR phase, the average recovery period is around 9 months, with a range of 1.9 to 32.9 months [25]. There is a lack of consensus in defining the partial clinical recovery phase (PCRP) in T1DM, and different interpretations have been proposed. One possible definition is when insulin requirement is less than 0.5 units/kg of body weight per day and HbA1c is below 7%. However, the gold standard for defining PCRP is a formula called the insulin dose-adjusted HbA1c (IDAA1c) formula [26]. The IDAA1c formula combines HbA1c (%) with 4 times the total daily dose of insulin (TDDI) in units/kg/24 hours, and a value lower than 9 indicates PCRP. This description is consistent with a stimulated C-peptide level exceeding 300 pM, and its validity has been confirmed in multiple extensive cohort studies [20, 26, 27]. Children who experience moderate to severe diabetic ketoacidosis at the time of T1D diagnosis are less likely to enter the partial recovery phase, and if they do, the duration of the phase is shorter [23]. In children under 2 years of age, PCRP is less frequent. This observation suggests a more accelerated and progressive destruction of β-cells, resulting in lower residual insulin secretion at the time of diagnosis [28]. Another study, although not replicable, discovered that men with T1D have a higher likelihood of experiencing PCRP and a longer duration of recovery compared to women with T1D [29]. Additionally, the type of insulin therapy and accurate blood sugar control with basal bolus or insulin pump may influence the duration of PCRP [30, 31].
The precise mechanisms underlying the partial clinical recovery phase (PCRP) in patients with T1DM remain largely unknown. However, several proposed mechanisms are thought to contribute to this period of relative β-cell recovery:
Temporary relief of glucose toxicity
Before diagnosis and initiation of insulin therapy, constant exposure to high blood glucose levels (referred to as “glucose toxicity”) can stress and exhaust β-cells. Improved glycaemic control through insulin therapy allows the β-cells to recover [32].
Improved insulin sensitivity
Prior to diagnosis, insulin secretion and insulin sensitivity gradually decrease in response to increasing levels of hyperglycaemia, described by Unger et al. as the “hyperglycaemia cycle”. Correcting hyperglycaemia through insulin therapy interrupts this cycle and restores insulin sensitivity. Studies using euglycaemic insulin clamp techniques have shown a reversal of insulin resistance in individuals newly diagnosed with T1DM who experience remission at 3 months after initiating insulin treatment [33].
Reduction in cytokines/inflammatory response in the islets (insulitis)
The honeymoon phase may have an immunological component. Lower levels of certain cytokines, such as interferon γ, interleukin-10, and interleukin-1R1, have been observed in individuals with partial remission. Additionally, the anti-inflammatory cytokine interleukin-1 receptor antagonist (IL1ra) is associated with higher stimulated C-peptide levels or improved β-cell function. These findings provide a basis for interventions aimed at modulating the immune response to control autoimmunity shortly after T1DM diagnosis [34, 35].
Increased glucose uptake in peripheral tissues
In individuals experiencing remission, there is evidence of significantly increased glucose transport into peripheral tissues (such as adipocytes) through insulin action. However, this effect disappears when the patient is no longer in the honeymoon phase.
Regeneration of β-cell mass
Animal studies conducted on NOD diabetic mice have shown that β-cell proliferation increases with islet inflammation during the progression of diabetes. This increased proliferation results in an expansion of “new” β-cells if autoimmunity is halted. It remains unclear whether this regeneration process occurs in humans. It is believed that the rapid recovery of islet function during partial remission is due to the recovery of previously exhausted β-cells that were not producing insulin but were present at the time of diagnosis [36, 37]. Table I provides a summary of biomarkers and their changes during the PCR phase.
Table I
Biomarkers and their alternations in the PCR phase
| Immunological and genetic factors involved in PCRP of T1DM patients | |||
|---|---|---|---|
| Type of biomarker | Name | Association/correlation pattern | Ref. No. |
| Cytokines | IL-6 | Positive | [38] |
| IL-10 | Positive | [39] | |
| IFN-γ | Negative | [40] | |
| CCL-3 | Positive | [38] | |
| CCL-4 | Irrelevant | [41] | |
| CCL-5 | Negative | [38] | |
| Molecules | CTLA-4 | Positive | [42] |
| Insulin | Positive | [43] | |
| PD-1 | Positive | [44, 45] | |
| PD-L1 | Positive | [44, 45] | |
| T cell subset | CD4+CD45RO+ | Positive | [46] |
| CD25+CD127hi | Negative | [46] | |
| Treg | CD39 | Positive | [47] |
| Treg apoptosis | Negative | [48, 49] | |
| B cell | Insulitis | Positive | [50] |
| Differentiation | Positive | [51] | |
| Gene | PTGER4 | Negative | [5] |
| Immunological and genetic factors involved in PCRP of T1DM patients | |||
| Immunological markers | Alternation pattern during the PCR phase | Ref. No. | |
| aTreg, Th17, Breg, Neutrophil, CCL-3, IL-6 | Increased at the beginning of the remission phase | [41, 52, 53] | |
| B cells, mononuclear, NK cells, CCL5, INF-γ, TGF-β, β-cell immunogenicity | Decreased at the beginning of the remission phase | [40, 41, 52-54] | |
| aTreg; CD4+CD25+CD127hi cells; CD4+CD45RO+ memory cells; Neutrophil; IL-10 | Positive correlation with the length of the remission phase | [39, 46, 52] | |
| NK cells; IL-4; IL-13; TNF-α | Negative correlation with the length of the remission phase | [52] | |
Factors affecting the PCR phase
The frequency and duration of the partial clinical recovery (PCR) phase in T1DM are influenced by various clinical and metabolic factors, which are partially dependent on the restoration of β-cell function [55]. Initially, it was hypothesised that this phase takes place due to the partial recovery of β-cells, resulting in the production of insulin and alterations in peripheral insulin resistance following the initiation of treatment by insulin [26]. Nevertheless, recent studies have indicated that these explanations are lacking [56, 57]. Presently, it is hypothesised that the honeymoon phase arises from the transient restoration of adaptive immune tolerance, but reliable biomarkers to reflect the attenuation of the autoimmune response and investigate β-cell proliferation are lacking [58]. In this context, it is postulated that an upsurge in the production of natural insulin takes place. Simultaneously, the initiation of insulin therapy leads to a reduction in the detrimental effects of glucotoxicity within the islets, potentially playing a role in the amelioration of residual β-cell function. Alongside the correction of glucotoxicity through exogenous insulin treatment, which allows β-cell rest [59], emerging evidence suggests that the normalisation of hyperglycaemia might preserve β-cell function through other mechanisms, including immune modulation [2, 56].
Recent research has highlighted the significance of B lymphocytes in the development of autoimmune diabetes, adding to the understanding of immunological factors [60, 61]. Specifically, studies have demonstrated that alterations in the frequency of marginal zone B cells (MZB), follicular B cells (FoB), and other cell subsets are associated with T1DM, and regulatory B cells (Breg) exhibit immunomodulatory effects [61]. Limited reports have indicated that changes in B-cell frequency are linked to the remission phase [52, 53]. In 2018, Fitas et al. [52] discovered a significant decrease in the absolute and relative abundance of B cells as the disease progressed, reaching its lowest level during the recovery phase. Another recent study revealed a substantial increase in Breg cells during the remission phase [53]. Additionally, the administration of the monoclonal anti-CD20 antibody rituximab as an intervention has shown promise in extending the duration of the remission phase up to 2 years [62]. Taken together, these discoveries indicate the participation of B lymphocytes in the remission phase of T1DM. Within the various subsets of B cells, the Breg subpopulation has gained significant attention due to its immunomodulatory properties, making it a focal point of research. Furthermore, other immune cells, including natural killer (NK) cells, monocytes, and neutrophils, may also contribute to the recovery phase of T1DM. In a study conducted by Fitas et al. [52], it was noted that there was a noteworthy reduction in the count of neutrophils and natural killer (NK) cells during the onset of T1DM. However, these cell populations exhibited a gradual recovery during the recovery phase. Additionally, the researchers observed a positive correlation between a lower percentage of NK cells and a higher percentage of neutrophils with the duration of the recovery phase. The frequency of specific T-cell subsets was also found to be closely linked to the stage of recovery, and it is suggested that this correlation may be partially attributed to effective glycaemic control. Moya et al. [46] conducted a study that revealed T1DM patients with a higher frequency of activated regulatory T cells (aTregs), CD45RO+ memory T cells, and CD25+ CD127hi cells experienced a prolonged recovery phase. However, this correlation was observed only when proper glycaemic control was maintained. Furthermore, a study from 2018 demonstrated dynamic changes in Treg cells, Th1 cells, and Tc1 cells during the recovery phase. Towards the end of the remission phase, there was a decrease in Treg cells and an increase in Th1 and Tc1 cells [52]. In a recent study involving newly diagnosed children with T1DM, a decrease in regulatory-memory T cells (mTregs) was observed after the onset of clinical symptoms. Conversely, a significant increase in aTregs and Th17 cells occurred during the first year, often coinciding with the recovery phase [53]. These findings suggest that the immune response during the recovery phase is influenced by the frequency and function of Treg cells, which undergo dynamic changes. Importantly, EP receptors, particularly EP4, are expressed by various immune cells, including T lymphocytes, macrophages, and dendritic cells. Prostaglandin E2 (PGE2) binds to EP4 receptors and exerts immunosuppressive effects by modulating the function of these crucial immune cells involved in immune responses [63, 64].
Prostaglandin E receptor 4 (PTGER4) gene and signalling pathways
The PTGER4 gene, also known as EP4 or EP4R, is located on chromosome 5p13.1 and consists of 7 exons. It encodes a transmembrane protein that functions as a receptor for prostaglandin E2 (PGE2) ligand [65]. PGE2, a key mediator in inflammation, exerts diverse effects in various tissues. It is the primary prostanoid released by cells in response to inflammatory stimuli [66, 67]. PGE2 is produced through the sequential metabolism of arachidonic acid by cyclooxygenase (COX) and prostaglandin E synthases (Fig. 2). Its biological functions are primarily mediated by 4 G protein-coupled receptors with 7 transmembrane domains: EP1, EP2, EP3, and EP4 [68]. Initially identified as a Gαs protein-coupled receptor, the EP4 receptor activates several downstream signalling pathways upon binding to its ligand PGE2. One of these pathways involves the stimulation of adenylate cyclase, leading to increased intracellular levels of cyclic adenosine monophosphate (cAMP) [69]. EP4 stimulation is associated with various functions, including vascular relaxation through the cAMP/protein kinase A (PKA)/endothelial nitric oxide synthase (eNOS) pathway [70], as well as angiogenesis via the cAMP/PKA Cµ pathway [71]. Phosphorylation of the transcription factor cAMP-responsive element-binding protein or CREB can be induced by various kinases, including protein kinase A or PKA and mitogen-activated protein kinases or MAP kinases [72, 73]. One of the important target genes of CREBP is the FcγRIIA gene, which plays a critical role in the response of neutrophils and monocytes to bacterial infections. PGE2, whether produced internally or introduced externally, activates CREBP by PKA in the PBL cell line differentiation, a process that relies on receptors of EP4 (74). In monocytes, activation of CREBP through the EP4 receptor leads to increased expression of the chemokine receptor CCR7 by binding to its promoter. This upregulation of CCR7 enhances the migration of monocytes [75]. On the other hand, cAMP can bind to the inducible cAMP early repressor (ICER) and negatively regulate the transcription of target genes. ICER, which is a truncated analogue of CREB lacking the trans-activation domain, suppresses the transcription of target genes by binding to cAMP response elements (CREs) in their promoters. For example, activation of the PGE2/EP4 signalling pathway can lead to ICER-mediated inhibition of retinoic acid dehydrogenase expression, resulting in decreased production and secretion of retinoic acid [76]. Another molecule involved in EP4 receptor signalling, independent of protein kinase A (PKA), is the cAMP-activated exchange protein (Epac). Epac-1 and Epac-2 function as guanine nucleotide exchange factors (GEFs) that mediate the communication between cAMP and members of the Ras superfamily, specifically Rho, Rac, and Ras. The signalling pathway regulated by Epac is widely involved in essential cellular functions, including cell proliferation, differentiation, migration, and inflammatory responses [77]. Additionally, the PKA and Epac signalling pathways can act synergistically in certain processes, such as the closure of the ductus arteriosus [78]. EP4 receptors in kidney podocytes can initiate COX-2 signalling through the AMP-activated protein kinase (AMPK) pathway, which is cAMP-dependent and PKA-independent [79]. Consequently, the PGE2/EP4 signalling axis induces several cAMP-dependent signalling pathways, such as PKA, AMPK, and Epac, that may work together or independently to mediate the impacts of the EP4 receptor mediated by Gαs [80, 81]. The C-terminal region of the EP4 receptor contains potential phosphorylation sites for PKA and G protein-coupled receptor kinases (GRKs), offering extra interaction sites for signalling molecules like EP4 receptor-associated proteins (EPRAP) or arrestins [66, 67]. Furthermore, the EP4 receptor possesses 2 N-glycosylation sites that appear to influence receptor surface expression [82]. Six serine residues in the long tail of the C-terminus of the EP4 receptor have been identified as potential targets for phosphorylation by PKA and GRK, thereby contributing to receptor desensitisation [83, 84]. Phosphorylation of the EP4 receptor leads to the binding of β-arrestin-1, which activates c-Src, initiating the activation of the epidermal growth factor receptor (EGFR) and downstream signalling through 3-phosphatidyl-inositol (PI3K) and Akt. This signalling pathway has been implicated in the regulation of metastasis process in colorectal cancer [85]. Additionally, the EP4 receptor can couple with pertussis toxin-sensitive inhibitory G-protein (Gαi), resulting in the activation of the PI3K/ERK signalling axis in HEK293 cells with increased expression of EP4 receptors [86, 87]. Stimulation of the EP4 receptor by PGE2 utilising the ERK signalling axis induces neurovascularisation in rats in vivo and promotes endothelial migration and tube formation in vitro [88]. The EP4 receptor-mediated protection in a murine model of cerebral ischaemia relies on subsequent activation of the Akt/eNOS downstream pathway [89].
Figure 2
A) EP receptors are coupled with different G proteins and accordingly activate different signalling pathways. B) Activation by binding to its cognate ligand, PGE2, leads to the activation of several downstream signalling pathways
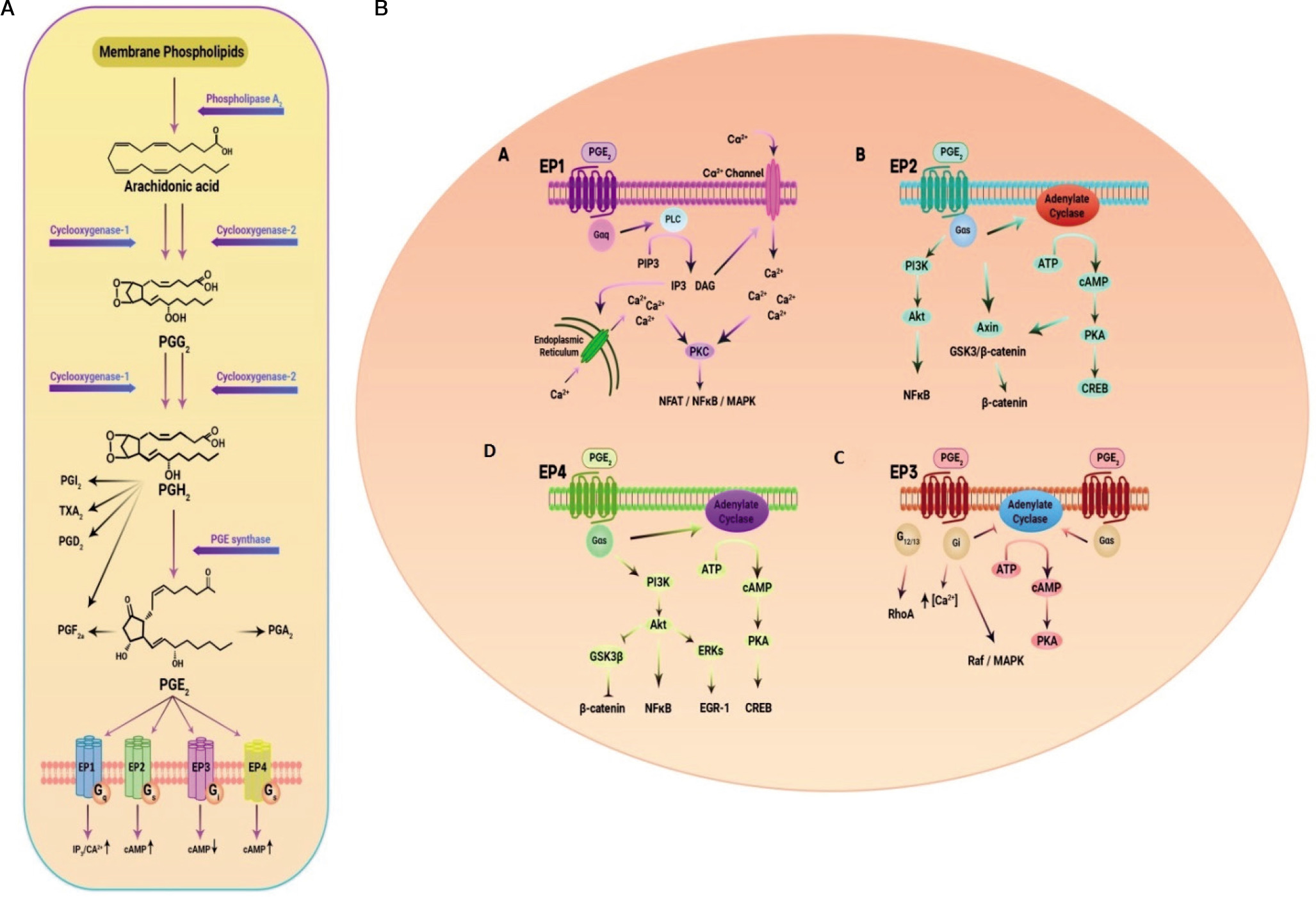
Moreover, decreasing the eosinophil effect by a selective EP4 receptor agonist has been demonstrated to be dependent on PI3K/PKC signalling rather than cAMP/PKA signalling [90]. Adding complexity to the EP4 receptor signalling pathway, the human macrophage’s EP4 receptor possesses a binding site for EPRAP within its extensive cytoplasmic tail [81]. EPRAP has been shown to stabilise the p105 subunit of NF-κB, preventing NF-κB activation and mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 (MEK/ERK1/2) signalling, leading to further inhibition of pro-inflammatory cytokine transcription (Fig. 2B) [91]. In human and murine macrophages, EP4 receptors exhibit an anti-inflammatory effect through G protein-independent signalling mechanisms and potentially decrease the immature B cells proliferation [81, 91, 92].
Association of EP4 with T1DM and subsequent complications
In individuals with T1DM, monocytes present in the bloodstream exhibit increased production of IL-6 and IL-1β, leading to the induction of Th1/Th17 cells [93]. IL-1 plays a crucial role in stimulating the synthesis of PGE2, which subsequently inhibits the activity of NLRP3, a member of the NLR family involved in regulating innate immune inflammatory responses by activating IL-1 and IL-28 cytokines [94]. However, PGE2 can also inhibit NLRP3 activation and reduce IL-1 production under specific homeostatic conditions, and signalling mediated by PGE2 receptors can exert anti-inflammatory effects in certain situations [95, 96]. Rahman et al. [97] demonstrated an increase in several inflammatory molecules, including PGE2, EP4, and IL-1β, just prior to the onset of T1DM. However, the IFN-I response was decreased, indicating an imbalance in intrinsic signalling. Although IL-1 is implicated in T1DM pathogenesis [98], blocking IL-1 signalling alone does not prevent the progression of T1DM [99]. IL-1 and IFN-I signals can counteract each other, and both signals are important for immune activation [100–103]. Activation of the PGE2/EP4 signalling pathway suppresses inflammation in pancreatic islets by promoting the transition of M1 macrophages to the M2 phenotype (thereby increasing the M1/M2 ratio) in patients with T2DM. This activation also protects β-cell function by modulating the inflammatory macrophages in the diabetic pancreas [104]. Indeed, islet inflammation and β-cell dysfunction are influenced by the infiltration of macrophages into pancreatic islets and the subsequent activation of pancreatic macrophages [105, 106]. Macrophages are characterised by 2 main phenotypes: the pro-inflammatory M1 phenotype and the anti-inflammatory M2 phenotype. It is possible for macrophages to switch from the M1 phenotype to the M2 phenotype, leading to enhanced insulin sensitivity [107]. In patients with type 2 diabetes mellitus (T2DM), activation of the EP4 pathway through PGE2 treatment or EP4 agonists can effectively suppress inflammation in adipose tissue associated with obesity and enhance insulin sensitivity. This effect is achieved by promoting the polarisation of macrophages toward the anti-inflammatory M2 phenotype, both in laboratory experiments (in vitro) and in living organisms (in vivo) [108]. In summary, in the context of the diabetic pancreas, the activation of EP4 receptors has been observed to enhance glucose-stimulated insulin secretion (GSIS) from pancreatic β-cells. This effect is achieved by inhibiting the inflammatory activation of macrophages. This switch in macrophage phenotype probably contributes to the overall effect. The exact role of PTGS (prostaglandin-endoperoxide synthase) enzymes in β-cells is not yet completely understood. However, substantial evidence supports the involvement of PTGS-2, among other factors, in the cytokine-induced damage to β-cells [109]. In non-obese diabetic (NOD) mice, the expression of PTGS-2 in β-cells disappears as diabetes progresses [110]. PTGS enzymes facilitate the conversion of prostaglandin endoperoxide, generated by the metabolism of arachidonic acid by PTGS enzymes, into a diverse array of prostanoids. These prostanoids include PGE2, PGD2, PGF2α, PGI2, and thromboxane A2-TXA2 [111]. PGE2, the primary product of PTGS in pancreatic tissue, exerts its effects on target cells through interaction with its 4 G protein-coupled receptor (GPCR) subtypes: PTGER1, PTGER2, PTGER3, and PTGER4, also known as EP1-EP4. These receptors are linked to diverse signalling pathways, including phosphoinoside hydrolysis and increased calcium levels for PTGER1, inhibition of adenylate cyclase (AC) for PTGER3, and activation of AC for PTGER2 and PTGER4 [112]. Human pancreatic islets have been reported to exhibit mRNA expression of all 4 subtypes of PTGERs [113]. In terms of pancreatic physiology, it has been demonstrated that PGE2 inhibits GSIS [114, 115], and this effect is probably mediated through its interaction with PTGER3 [116].
A recent study by Wu et al. [117] unveiled the presence of specific G protein-coupled receptors (GPCRs) localised in the primary cilia of α- and β-cells within the islets of Langerhans, which play a role in regulating the secretion of insulin and glucagon. The primary cilium is a specialised organelle composed of membranes and microtubules, which extends from the apical surface of cells and contains a high concentration of specialised GPCRs [118]. These cilia are found in both α- and β-cells of pancreatic islets in mice and humans [119], and emerging evidence suggests that diabetes development in individuals with ciliopathies is associated with impaired insulin secretion [120, 121]. Furthermore, studies have linked dysregulation of cilia-related genes to an increased risk of both T1DM and T2DM [122–124]. Animal models have demonstrated that α- and β-cells with fewer cilia exhibit incomplete glucose-regulated insulin secretion [122, 125, 126]. Additionally, specific knockout mice lacking the ciliary core gene Ift88 or the BBSome complex member Bbs4, both essential for ciliary signalling and trafficking, showed impaired glucose-stimulated insulin secretion (GSIS) [122, 127–129]. Wu et al. focused on assessing the role of ciliary GPCRs as regulators of insulin and glucagon secretion in islets. They identified GPCRs, including FFAR4 and PTGER4, localised within the cilia of native α- and β-cells, which regulate insulin and glucagon secretion in response to pharmacological agonists through the activation of localised ciliary cAMP signalling. Activation of the FFAR4 receptor by its natural ligand, ω-3 fatty acid, within the cilia of α- and β-cells leads to increased cAMP levels and subsequent activation of downstream effectors such as EPAC and PKA. This activation ultimately stimulates the secretion of glucagon from β-cells and insulin from β-cells. Moreover, stimulation of FFAR4 can induce insulin and glucagon secretion through the activation of phospholipase C (PLC), resulting in an increase in intracellular calcium levels [130]. Among the 4 prostaglandin E2 receptors (EP1-EP4), only EP4/PTGER4 is localised in the cilia [131] and is primarily associated with Gαs and cAMP production. Previous studies have demonstrated the protective role of EP4 in β-cell survival and proliferation, while EP3 may have opposing effects [132]. The presence of inflammatory receptors like PTGER4 may link local pancreatic inflammatory responses or more general systemic inflammatory responses to the efficiency of insulin secretion by the islets. High levels of systemically delivered PGE2 have been shown to reduce insulin secretion [133, 134], but the specific effects of PGE2 on β-cell proliferation, survival, and insulin secretion, as well as its effects outside of β-cells, remain to be fully understood. Previous studies have suggested that PGE2 may inhibit insulin secretion through a different PGE2 receptor, PTGER3, but a consensus on the control of PGE2 on insulin secretion has yet to be established [133]. Wu et al. demonstrated that an agonist for PTGER4 increases cAMP levels through ciliary signalling, leading to a positive effect on insulin release [117].
Diabetic nephropathy
Among the complications associated with diabetes, diabetic nephropathy (DN) is considered a major cause of morbidity and mortality, as well as the leading cause of end-stage kidney disease in developed countries [135]. DN presents with renal impairment resulting from the excessive accumulation of extracellular matrix proteins on the glomerular surface, leading to the expansion of mesangial cells and thickening of the glomerular basement membrane (GBM). The underlying cause of these alterations is the abnormal activation of remaining renal cells, which leads to an elevated production of transforming growth factor β1 [136, 137].
Berberine (BBR) is a type of isoquinoline alkaloid that is obtained from the rhizome of coptidis and the cortex of Phellodendron plants. This compound has garnered significant interest due to its diverse therapeutic properties in managing diabetes and its related complications. Some of these beneficial effects include its ability to lower blood glucose levels, reduce lipid levels, enhance insulin sensitivity, inhibit the polyol pathway, and exhibit antioxidant activity. As a result of these properties, berberine has become widely utilised as a treatment option [138–140]. In China, BBR is commonly employed to lower blood glucose levels in patients with T2DM and has been found to be as effective as metformin, a commonly prescribed drug for T2DM treatment [141]. Notably, BBR has demonstrated its ability to reduce blood glucose levels and alleviate renal dysfunction and mesangial matrix expansion in diabetic rats induced by streptozotocin (STZ) [142, 143]. Recent studies have further revealed the renoprotective effects of BBR in diabetic rats, showing that early initiation of BBR therapy significantly delays the progression of diabetic nephropathy (DN). This renoprotective effect is attributed, in part, to the inhibition of the RhoA/ROCK signalling pathway, which helps mitigate renal inflammation and fibronectin accumulation in the kidneys of diabetic rats [144]. Additionally, BBR has shown dose-dependent nephroprotective effects against kidney injury induced by cisplatin (CP). It achieves this by reducing the expression of CYP2E1 and inhibiting the pro-inflammatory NF-κB pathway [145]. Yang et al. demonstrated that BBR exhibits different therapeutic effects at different stages of DN development, with its optimal performance observed at the sixth week. The study also suggested that BBR exerts renoprotective effects in different stages of DN through the PGE2/EP4/Gαs/AC/cAMP signalling pathway in STZ-induced DN rats [146]. Wang et al. uncovered an unexpected but important role of the COX2/PGE2/PTGER4 signalling axis in macrophages of T1DM. In contrast to the detrimental effects observed with increased COX-2 expression in intrinsic kidney cells, this signalling pathway mitigates the progression of diabetic kidney disease [147]. Previous studies have highlighted the significance of this pathway in macrophage polarisation towards an M2 phenotype and have revealed that EP4 activation inhibits the activation of pro-inflammatory cytokines and NLRP3 inflammasome stimulation in macrophages [148]. The impact of COX-2 in kidney injury is contingent upon multiple factors, such as the origin of COX-2, mechanisms underlying renal injury, expression and subtype of PGE2 receptor, and the duration of COX-2 inhibition [95, 149, 150]. A study conducted by Riyaz Mohammad et al. discovered that the administration of the EP4 receptor agonist ONO-AE1-329 for 12 weeks exacerbated albuminuria and fibrosis in mice [151]. The kidneys of mice treated with the EP4 agonist showed increased expression of inflammatory cytokines induced by diabetes, such as TNF-α and TGFβ1. The study data indicated that the EP4 agonist induces glomerulosclerosis and interstitial fibrosis through the involvement of IL-6, which acts as a new player in the PGE2/EP4 signalling pathway. Activation or overexpression of the EP4 receptor also led to increased membrane accumulation of AQP2 (aquaporin-2) in a murine inner medullary collecting duct cell line (IMCD3) that was transfected with the AQP2 gene. This effect was primarily mediated through cAMP/PKA and kinase signalling pathways regulated by extracellular signals [152]. Under common physiological circumstances, the EP4 receptor plays a critical role in governing urine concentration in renal collecting ducts. Concentration of urine is vital for keeping water balance within the body and is primarily controlled by the antidiuretic hormone arginine vasopressin (AVP).
Macrovascular complications
Both T1DM and T2DM exhibit macrovascular complications that heighten the likelihood of experiencing myocardial infarction, stroke, and peripheral vascular disease. These complications mainly arise due to an elevated presence of atherosclerosis. The precise mechanisms through which diabetes contributes to the progression of atherosclerosis remain incompletely understood. T1DM and T2DM are characterised by heightened levels of blood glucose and frequently coincide with an increase in inflammatory markers. Individuals with T1DM often exhibit additional cardiovascular risk factors such as dyslipidaemia and insulin resistance. Moreover, other risk factors like hypertension, smoking, and nephropathy, if present, can further contribute to the amplified risk of cardiovascular disease, mirroring the situation in individuals without diabetes. It has been demonstrated that keeping tight control over blood glucose levels can lessen the risk of future cardiovascular complications in young patients with T1DM [86, 156]. The precise understanding of how increased glucose, lipids, inflammation, and other factors linked to T1DM contribute to the risk of cardiovascular disease remains incomplete. Myeloid cells derived from both humans and animal models of diabetes commonly demonstrate enhanced activation, resulting in heightened expression of chemokines and cytokines. Additionally, there is often an expansion of Th17 cells observed [93, 157–161]. Furthermore, studies have demonstrated that diabetes contributes to heightened inflammatory myelopoiesis in animal models [162]. This suggests that the inflammatory state of myeloid cells in diabetes may partially account for the impact of diabetes on atherosclerosis. One potential factor that could enhance the inflammatory activity of myeloid cells in diabetes is prostaglandin E2 (PGE2). Studies have shown that PGE2 can induce the upregulation of multiple processes and inflammatory factors in myeloid cells. These include IL-6 and the chemokine receptor CCR7 [75, 163, 164]. Conversely, PGE2 inhibits factors such as TNF-α, CCL5, and activation of inflammasome [95, 163, 165]. The diverse effects of PGE2 can be attributed to the presence of 4 G protein-coupled PGE2 receptors (EP1-EP4), with EP4 specifically associated with inflammation [166]. Activation of EP4 by PGE2 triggers multiple intracellular signalling events, such as increased cAMP levels through adenylate cyclase (AC) activation and subsequent activation of the transcription factor CREBP, as well as phosphatidylinositol 3-kinase (PI3K) activation [167]. In macrophages, EP4 activation also inhibits NF-κB following toll-like receptor 4 (TLR4) activation by lipopolysaccharide [91]. PGE2 synthesis is induced by numerous inflammatory mediators and is frequently raised during states of severe inflammation [168]. Consequently, research has reported elevated plasma or urinary levels of PGE2 in patients with T1DM, although some have shown no significant difference [169, 170]. PGE2 has been implicated as a potential mediator of certain diabetes-related complications [170–173]. For instance, administration of an EP4 agonist worsened renal fibrosis in diabetic mice induced by streptozotocin and increased the expression of inflammatory cytokines like IL-6 [151]. Vallerie et al. [174] demonstrated that in a mouse model of T1DM, the PGE2-EP4 signalling axis was upregulated in myeloid cells, which was necessary for the diabetes-induced stimulatory effect on IL-6, while it had a mitigating effect on TNF-α and did not moderate the accelerated atherogenesis associated with diabetes. An interesting discovery suggests that while PGE2 primarily induces fibrous tissue in new bone formation, the use of an EP4 agonist promotes well-connected and adequately calcified trabeculae, resulting in stronger new bone [177]. However, the systemic administration of PGE2 is limited due to its unwelcome effects, such as hypotension, diarrhoea, uterine contractions, and intestinal epithelium thickening. Consequently, the targeted use of a specific PGE2/EP4 receptor agonist has shown promising results in accelerating the recovery of devascularised sternum, significantly reducing the incidence of sternum-related complications, even in diabetic rat models. This approach could potentially expand the utilisation of bilateral internal thoracic arteries (BITAs) in coronary artery bypass surgery for high-risk diabetic patients, leading to excellent long-term outcomes. Moreover, the rapid sternal recovery shortens hospital stays, reduces healthcare costs, and facilitates the early resumption of work and social activities for patients.
Microvascular complications and diabetic retinopathy
Neovascularisation (NV) is a vital process during embryonic development, which can also be reactivated in adulthood to support tissue growth, wound healing, and the menstrual cycle. However, when blood vessel sprouting becomes excessive or uncontrolled, it can contribute to the development of various diseases [178]. Proliferative vitreoretinal neovascularisation, which includes conditions such as proliferative diabetic retinopathy (PDR), retinopathy of prematurity (ROP), and retinal vein occlusion, is a significant contributor to visual impairment and blindness on a global scale, specifically in ocular disorders [179–182]. Prostanoids, a family of bioactive lipids derived from arachidonic acid by the action of COX-1 and COX-2 enzymes, play a critical role as growth factor inducers and potent proangiogenic mediators [183]. Studies have demonstrated that in hepatocellular carcinoma (HCC) cells, PGE2 plays a notable role in stimulating cell growth and invasion. This effect is achieved through the activation of the classical EP4/Gαs/AC/cAMP/PKA signalling pathway [184]. Furthermore, prior research findings have indicated that PGE2 activates the EP4 receptor, resulting in the assembly of the EP4/β-arrestin1/Src complex. This complex, in turn, transactivates the epidermal growth factor receptor (EGFR) [185–187]. This activation cascade involves the binding of growth factor receptor-bound protein 2 (Grb2) to EGFR, which then recruits Grb2-associated binding protein 1 (Gab1), ultimately enhancing or prolonging the activity of the Akt signalling pathway through the recruitment of the p85 subunit of PI3K [188, 189]. Similarly, Xie et al. showed that disruption of the PGE2/EP4 signalling pathway is implicated in reducing the pathogenesis of retinal neovascularisation [190]. Activation of the EP4 receptor by PGE2 leads to EGFR transactivation and the utilisation of Gab1, ultimately promoting cell proliferation in a model of retinopathy of prematurity (ROP) through the Akt signalling pathway.
Conclusions
The PCR phase, which serves as a clinical treatment phase, offers a valuable human model for studying the protection of β-cells and immune modulation. A comprehensive understanding of the underlying mechanisms in this phase could form a crucial foundation for inhibiting the autoimmune response and potentially developing clinical treatments for T1DM. It is worth noting, however, that β-cell regeneration can have both positive and negative effects. On one hand, it increases the production of endogenous insulin, but on the other hand, it induces the release of autoantigens, perpetuating a cycle of regeneration and autoimmunity. Nevertheless, the recovery phase has garnered significant interest as a potential target for therapeutic interventions in T1DM, aiming to preserve the remaining β-cells and identify individuals at low risk of long-term diabetes-related complications. Consequently, the identification of reliable and specific biomarkers for the PCR phase is crucial to identify at-risk individuals, monitor this phase more effectively, and implement appropriate interventions in the future. Although the exact mechanisms underlying PCR remain unclear, considering that genetic factors primarily contribute to the development of T1DM, it is plausible that there are also predisposing genes associated with entering the PCR phase. Notably, the discovery of an association between a variant of the EP4 receptor gene and modulation of the PCR phase suggests that signalling pathways triggered by the EP4 receptor could be involved. Given the role of different signalling pathways induced by PGE2/EP4 in regulating the functions of immune cells implicated in T1DM pathogenesis, particularly during the PCR phase, and the impact of immune molecules associated with this phase on the aforementioned axis, it is reasonable to hypothesise that the PGE2/EP4 axis plays significant roles in PCR. However, to validate this theory, several considerations should be addressed. Firstly, a more precise definition of the PCR phase is required to accurately determine its parameters. Secondly, research involving larger sample sizes should include comprehensive analysis of genetic variants of the EP4 gene and, if feasible, other important genes involved in the associated signalling axis. Lastly, functional investigations are necessary to confirm and establish the role of the EP4 gene and its associated signalling pathways in the PCR phase.









