
Hypoparathyroidism (HPT) is an uncommon endocrine disorder in children characterized by hypocalcemia, hyperphosphatemia, and absent or low levels of parathyroid hormone (PTH). It is caused by inherited or acquired defects involving the synthesis or secretion of PTH, resistance to PTH action, or inappropriate regulation of PTH by calcium-sensing receptor (CaSR) [1]. In adults, the usual cause of HPT is the inadvertent damage or removal of the parathyroid glands during neck surgery. In children, however, the genetic etiologies predominate [1]. Pediatric HPT may be isolated or syndromic i.e. associated with non-parathyroid related dysmorphisms or disturbances. Several syndromes, such as DiGeorge syndrome, HDR (hypoparathyroidism, sensorineural deafness, and renal dysplasia) syndrome, HRD (hypoparathyroidism, retardation, and dysmorphism) syndrome, KSS (Kearns-Sayre syndrome), may have associated HPT [1]. In all cases of non-surgical causes, a systematic clinical and genetically driven approach is necessary to reach the exact diagnosis of HPT [1]. Non-genetic autoimmune HPT, whether isolated or as part of the autoimmune polyglandular syndrome type 1 (APS-1) is extremely rare in children and is often a diagnosis of exclusion [1].
Jacobsen syndrome (JS) is a rare contiguous gene syndrome caused by partial deletion of the long arm of chromo-some 11. It is characterized by growth and psychomotor retardation, distinct facial features, and thrombocytopenia or pancytopenia [2]. Additional features include abnormalities of the heart, kidney, gastrointestinal tract, genitalia, central nervous system, skeleton, ocular, hearing, and immune system. Endocrine manifestations such as isolated hypogonadism, multiple pituitary hormone deficiencies (MPHD), and isolated growth hormone and thyrotropin deficiency have also been described in JS [2, 3]. The hormonal disturbances in patients with ring chromosome 11 (RC11) that may have overlapping clinical manifestations with JS include precocious puberty, hypothyroidism, type 2 diabetes (T2D), and polycystic ovary syndrome (PCOS) [4]. However, HPT has never been reported previously in patients with either JS or RC11.
The conventional treatment of chronic HPT with calcium and vitamin D analogs poses a significant risk of hypercalciuria, nephrocalcinosis, and renal impairment [5]. Patients often require additional therapies such as thiazide diuretics and phosphate binders in order to prevent long-term complications [5]. Although replacement therapy with recombinant human parathyroid hormone (rhPTH1–34) has physiological benefits, it is rarely used in pediatric practice due to the FDA’s black box warning [5]. In this report, we describe our decade-long experience of treating HPT with rhPTH1–34 in a boy with JS.
Case report
Approval for the case report was obtained from the Departmental Review Board. Informed consent was obtained from the parents. A 5½-year-old boy presented with several episodes of stiffening of limbs, abnormal posturing of the body, and intermittent laryngospasms for the past 2 days. Similar episodes occurred 6 months before. He was the second child of nonconsanguineous parents. There was no family history of seizures, hypocalcemic disorder, intellectual disability, apparent congenital malformations, or congenital heart disease. The motor development was normal, but personal social and language milestones were delayed. Parents also reported learning difficulties.
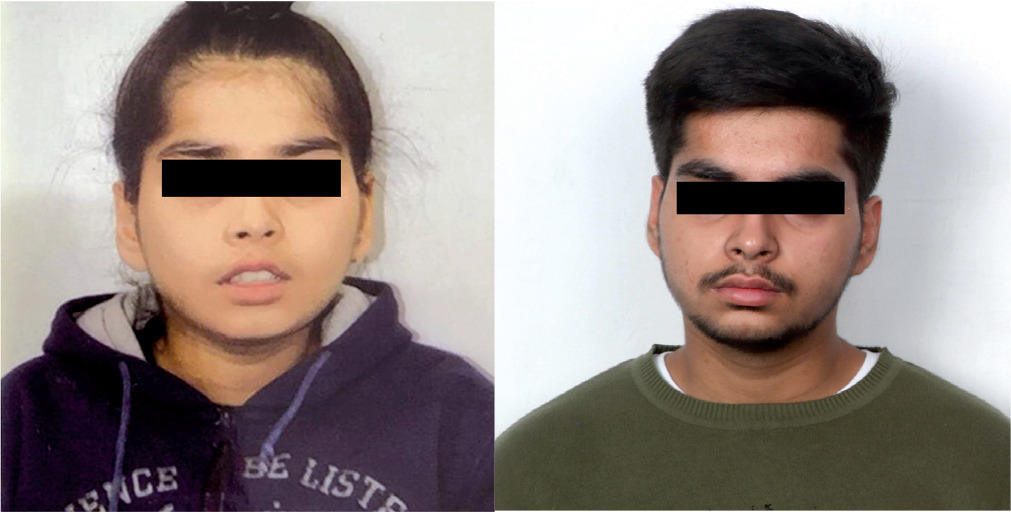
Physical examination showed a weight of 23.8 kg (1.41 z-score, World Health Organization [WHO] growth charts), height 119 cm (1.36 z-score, WHO growth charts), and head circumference of 52.0 cm (0.8 z-score, WHO growth charts). The parental heights were 186.6 cm and 177.8 cm, and his target height range was 180.2–196.2 cm. He had low set ears, hypertelorism, downslanting palpebral fissures, broad nasal bridge, and downturned corners of the mouth (Fig. 1). The systemic examination was unremarkable.
Figure 1
Clinical photograph showing facial features of the patient at 5.5 (A) and 16.5 years (B) of age. Consent for publication taken from parents and patient
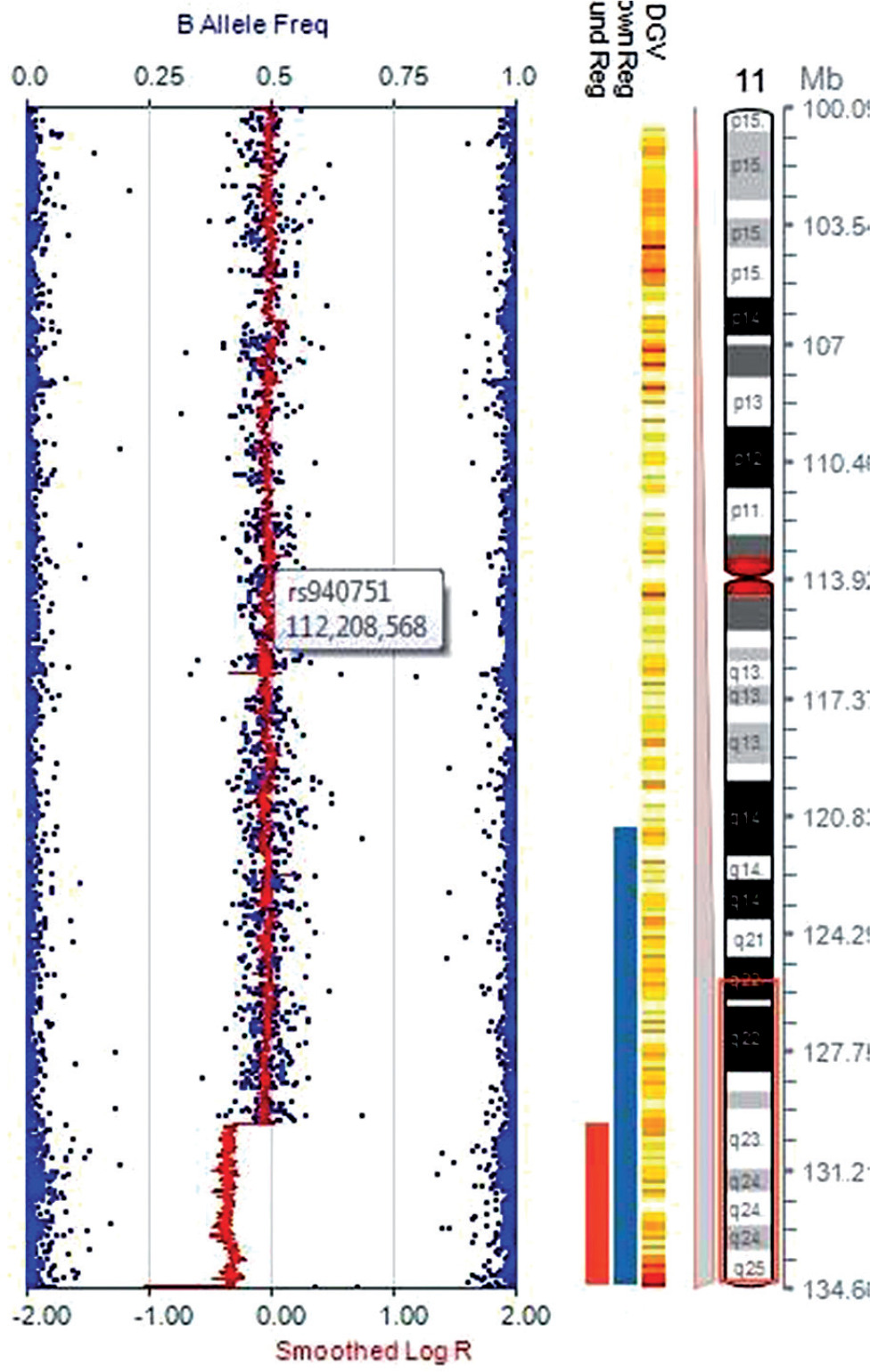
Investigations revealed low ionized calcium (0.2 mmol/l, normal range 1.1–1.35 mmol/l), low total serum calcium (4.4 mg/dl, normal range 9.6–10.6 mg/dl), high serum phosphate (9.3 mg/dl, normal range 3.7–5.4 mg/dl), and normal serum magnesium (1.2 mmol/l, normal range 0.7–1 mmol/l). Serum PTH was low (1.4 pg/ml, normal range 10–65 pg/ml). The hypocalcemic symptoms improved with intravenous calcium infusion, and he was discharged on oral calcium and calcitriol. A provisional diagnosis of syndromic HPT was considered. Targeted next generation sequencing (NGS) with 80–100X read depth did not reveal any mutations in HPT-associated genes such as PTH, TBX1, GATA3, GATA3, GCM2, and TBCE. Chromosomal micro-array analysis (CMA) showed a heterozygous deletion of 4.7 Mb in the cytoband 11q24.3q25 encompassing approximately 20 genes, including JAM3 and NTM genes (Fig. 2). Considering the facial phenotype and intellectual disability, a diagnosis of partial JS was made as patients with such observed deletion overlap with JS. In addition, the sample also showed a duplication of 770 kb in the cytoband 1p36.13 encompassing several genes (SDHB, PADI2, PAD1, PADI3, PADI4, PADI6, RCC2, ARHGEF10L, and ACTL8). However, this copy number variant (CNV) was classified as a variant of uncertain significance (VUS).
Figure 2
Chromosomal microarray analysis using Illumina Human CytoSNP-12 microarray chip, showing a deletion of chromosome 11q24.3q25 cytoband region
Magnetic resonance imaging of the brain was normal. The IQ assessment showed borderline deficiency in intelligence with an IQ score of 74. The initial urine calcium creatinine ratio was 0.04, and the renal ultra-sonogram was normal. He required high doses of oral calcium during the first year of follow-up, allowing him to be symptoms free despite low serum calcium levels (Table I). During the second year, the urinary calcium creatinine ratio increased to 0.35, and renal ultrasonography showed concretions in the kidneys. He was then started on Teriparatide (Forteo; Eli Lilly, Indianapolis, IN, USA). Adjustments of doses of rhPTH1–34 and calcium were done to maintain serum calcium in low normal ranges (Table I). Oral calcium was stopped after 8 years. A detailed assessment was carried out at the end of 10 years of rhPTH1–34 therapy. The serum bone specific alkaline phosphatase (ALP) was 103.33 U/l (normal range, 13–111 U/l), insulin-like growth factor-1 (IGF-1) 462.0 ng/ml (normal range, 220-972 ng/ml), IGF binding protein-3 6.39 μg/ml (normal range, 3.3–10.0 μg/ml), osteocalcin 139.2 ng/ml (normal range, 30.1–186.9 ng/ml), thyroxine 7.29 μg/dl (normal range, 4.8–12.7 μg/dl), thyrotropin 2.44 μIU/ml (normal range, 0.27–4.2 μIU/ml), cortisol AM 234.6 nmol/l (normal range, 171– 536 nmol/l), fasting blood glucose 76 mg/dl (normal, < 100 mg/dl) and glycated hemoglobin 5.7% (normal range, 4.8–5.7%). Serum PTH was < 0.23 pg/ml. No platelet abnormality was detected on hemogram, and the renal function was within the normal range. The whole-body Tc99m MDP bone scan showed no abnormal osteoblastic activity anywhere in the skeleton. The fan-beam dual X-ray absorptiometry scan of the whole body showed a subtotal BMD of 1.026 g/cm2 (0.7 z-score).
Table I
Mean values of various biochemical parameters monitored during follow up
Discussion
Patients with JS show considerable variability in clinical features [2]. Our patient had some of the typical features, such as facial dysmorphism and intellectual disability. However, other symptoms such as Paris-Trousseau syndrome seen in more than 90% of cases, and systemic malformations were absent. Patients with JS may also show clinical overlap with RC11 syndrome, which, in addition to other genetic alterations, may have terminal deletion in one or both arms of chromosome 11, similar to JS [4]. Jacobsen syndrome has been reported in several patients with RC11, and the pathogenesis of several metabolic alterations in both syndromes is speculated to be due to the involvement of gene(s) located on chromosome 11 [4]. In our patient, the presence of NTM gene deletion explains intellectual disability as the NTM gene is associated with the development of the nervous system [2]. However, no cardiac malformations were detected despite the presence of the JAM3 gene mutation, usually considered a candidate gene for the cardiac phenotype [2, 6]. Interestingly, JAM3 gene deletion, one of the 4 genes (FLI1, ETS1, NFRKB, and JAM3) related to thrombocytopenia did not manifest with Paris-Trousseau syndrome in our patient, similar to some previous cases [6, 7]. The partial expression of the JS phenotype in our patient may be related to the size of terminal deletion, as the most severe phenotype is usually seen in patients with the largest deletions [2]. The minimal region required for expressing the JS phenotype spans approximately 14 Mb compared to only 4.7 Mb in our patient [2]. The presence of duplication of 770 kb in the cytoband 1p36.13 in our patient classified as VUS has been reported in normal healthy persons. However, the phenotypic spectrum of 1p36 duplication may include developmental delay, mild facial dysmorphism, and neurological, cardiac, and skeletal anomalies [8].
Several endocrine manifestations such as T2D, PCOS, isolated, or MPHD are described in patients with JS and RC11 [2–4]. However, there are no previous reports of HPT in patients with either JS or RC11. The young age at diagnosis and presence of facial dysmorphism suggested a non-autoimmune and non-mitochondrial syndromic HPT in our patient. However, the clinical phenotype was not consistent with genetic syndromes usually associated with HPT such as DiGeorge, CHARGE, HRD, HDR, and KSS syndromes [1]. Additionally, the CMA did not identify any of the causative CNVs implicated in the listed syndromes, and the targeted NGS also did not find any mutations in the usual HPT-associated genes. However, we did not test for mutations in CaSR and the autoimmune regulator (AIRE) gene, which are extremely rare causes of sporadic HPT in children. Patients with sporadic or autosomal dominant hypocalcemia caused by loss-of-function mutations in CaSR characteristically have hypercalciuria, which in our patient appeared only after initiation of calcium therapy. APS-1 caused by AIRE gene mutations usually manifests with chronic mucocutaneous candidiasis (CMC) in infancy followed by HPT and Addison’s disease in later childhood. The clinical diagnosis of APS-1 requires the presence of the classic triad of symptoms, although patients with single manifestations have been described, which invariably is CMC and not HPT [9]. The characteristic manifestations of KSS, such as progressive external ophthalmoplegia, retinitis pigmentosa, and short stature were not seen in our patient. The cause of HPT, thus, remains idiopathic in our patient.
A highlight of our report is the successful long-term use of rhPTH1–34 for the usually challenging management of chronic HPT in children. The decision to start rhPTH1–34 in our patient was taken in view of hypercalciuria and renal concretions. However, fluctuations in serum calcium concentrations continued, and titration of oral calcium intake was required for several years. Such an observation is similar to the experience with HPT patients on long-term rhPTH1–34 therapy at other centers [10, 11]. A significant finding in our patient was the reduction in hypercalciuria and absence of nephrocalcinosis during the last 3 years of therapy. Similar observations in previous studies probably indicate the beneficial effects of the presence of PTH in renal tubules or reduced oral calcium intake [1, 5, 10, 11]. Another notable feature was a gradual reduction of rhPTH1–34 dose per kg body weight from 0.90 during the 6th year to 0.26 during the 10th year of therapy, although serum PTH concentrations remained low. The mean daily PTH doses of 0.75 ±0.15 μg/kg and 0.68 ±0.23 μg/kg used in previous long-term studies were probably due to the inclusion of cases of genetic HPT, which require higher dosages [10, 11]. Several recent studies have reported experience with rhPTH1–34 and recommend the use of rhPTH1–34 in adults as well as children for management of chronic HPT [10–14]. A recent review that included short and long-term experience on the use of rhPTH1–34 in 70 children suggested rhPTH1–34 to be an effective therapy for HPT that improves metabolic control and quality of life and is associated with better renal outcome in children with HPT [5]. Other reassuring features in our patient were a normal BMD after 10 years of PTH therapy, probably indicating a normal trajectory of bone accrual, and the attainment of height within the genetic potential.
The use of rhPTH1–34 is prohibited in children with open epiphyses due to concerns of osteosarcoma reported in animal studies that led to black box warning by the FDA [5]. We monitored bone turnover by estimation of serum ALP at each clinic visit in addition to specific enquiries about any bone-related symptoms. A detailed assessment at completion of 10 years of therapy did not indicate increased bone turnover, an observation consistent with several reports of short- and long-term rhPTH1–34 use in children [5, 10, 11]. Combined data from pediatric and adult studies accumulated over the last 15 years of rhPTH1–34 use is also reassuring and does not indicate an increased risk of osteosarcoma [10, 11]. In view of several disadvantages of conventional treatment of HPT in children, such as poor metabolic control, repeated emergency visits, poor quality of life, nephrocalcinosis, requirement of additional therapies, and renal damage, the use of rhPTH1–34 is probably justified despite the theoretical risk of osteosarcoma [5, 15]. However, continued surveillance for bone neoplasms is mandatory in all patients on treatment with rhPTH.









