
Introduction
Alterations in DNA methylation pattern are proven to have an impact on cancerogenesis. Novel developments in this field have heightened the need to establish the involvement of drugs used in cancer treatment in the epigenome. In the light of recent discoveries, epigenetic processes, including DNA methylation, may affect regulation of crucial genes implicated in drug response and drug targets [1]. Current experimental evidence has demonstrated that an active DNA demethylation process can affect DNA methylation pattern [2]. Tamoxifen, as a leading drug, which belongs to selective estrogen receptor modulators (SERMs), has been in clinical use for treatment and prevention of hormone-dependent breast cancer for nearly 40 years. Since its discovery, the use of tamoxifen has evolved by embracing its ability to act like estrogen in association with G-protein coupled receptors or antiestrogen in association to estrogen nuclear receptors [3, 4]. Given tamoxifen’s facility to act like an agonist or antagonist of estrogen receptors [5], its pharmacological activity may be more complex than just control estrogen activity. Thus, the role of this agent may be potentially involved with other factors or processes which occur in cells.
Active demethylation processes are essential during embryonic development and cell differentiation, although they can be modulated by environmental changes (e.g. after chemotherapy), which may manifest as random mutations [6]. Epigenetic modifications in the human genome may affect gene expression, particularly tumor suppressor genes, genes involved in apoptosis regulation and oncogenes [7], as well estrogen-dependent genes [8]. The level of epigenetic marks is flexible and mostly is associated with the response to hypomethylating factors.
DNA methylation and demethylation
The level of gene expression in cells is often conditioned by various epigenetic modifications. The most fundamental and widely described epigenetic alteration is DNA methylation of the cytosine residue, which in turn leads to formation of 5-methylcytosine (5-mC), the fifth DNA base [9]. Such terminology reflects the relative abundance of 5-mC content in the human genome: approximately 4% of cytosine residues are methylated [10]. DNA methylation is a physiological phenomenon, which takes part in cellular development and differentiation; however, it also plays a crucial role in the ageing process and carcinogenesis [11]. Vertebrate genomes are generally methylated in CpG islands, which are located at promoter regions of genes. The presence of multiple methylated CpG islands of promoters causes gene silencing [12]. Apart from DNA methylation, the 5-mC level is dependent on active DNA demethylation, which is being currently explored. Active DNA demethylation involves TET, and possibly AID proteins [13]. TET (ten-eleven-translocation) proteins are 2-oxoglutarate dependent dioxygenases (2-OGDDs), and the process catalyzed by these enzymes requires 2-oxoglutarate as a co-substrate, and Fe2+ and ascorbate as co-factors. Their catalytic function involves oxidation of 5-mC to 5-hydroxymethylcytosine (5-hmC), and then to 5-formylcytosine (5-fC), and 5-carboxylcytosine (5-caC). AID protein is an activation-induced deaminase which is implicated in deamination of 5-mC to thymine, and possibly 5-hmC to 5-hydroxymethyluracil (5-hmU) [14]. Additionally, TET enzymes could generate 5-hmU from thymine [15]. Cytosine modifications can be subsequently excised by TDG (thymine DNA glycosylase) or SMUG (single-strand selective monofunctional uracil-DNA glycosylase1) in the base excision repair system (BER) [16] (Fig. 1). DNA glycosylases replace modified bases with cytosines, which terminates in demethylated DNA formation [17]. The main role of TET proteins is preserving the fidelity of DNA methylation/demethylation at different stages of cellular development and differentiation. Recent research in this field has revealed that the differences in TET expression are already visible in oocytes and zygotes: from the highest expression of TET3 to intermediate of TET2, and hardly observed for TET1. Moreover, TET expression changes dramatically during further cell development: at the 4-cell stage TET1 and TET2 are expressed at a higher level than TET3, and this tendency remains at the same level until blastocyst formation [18].
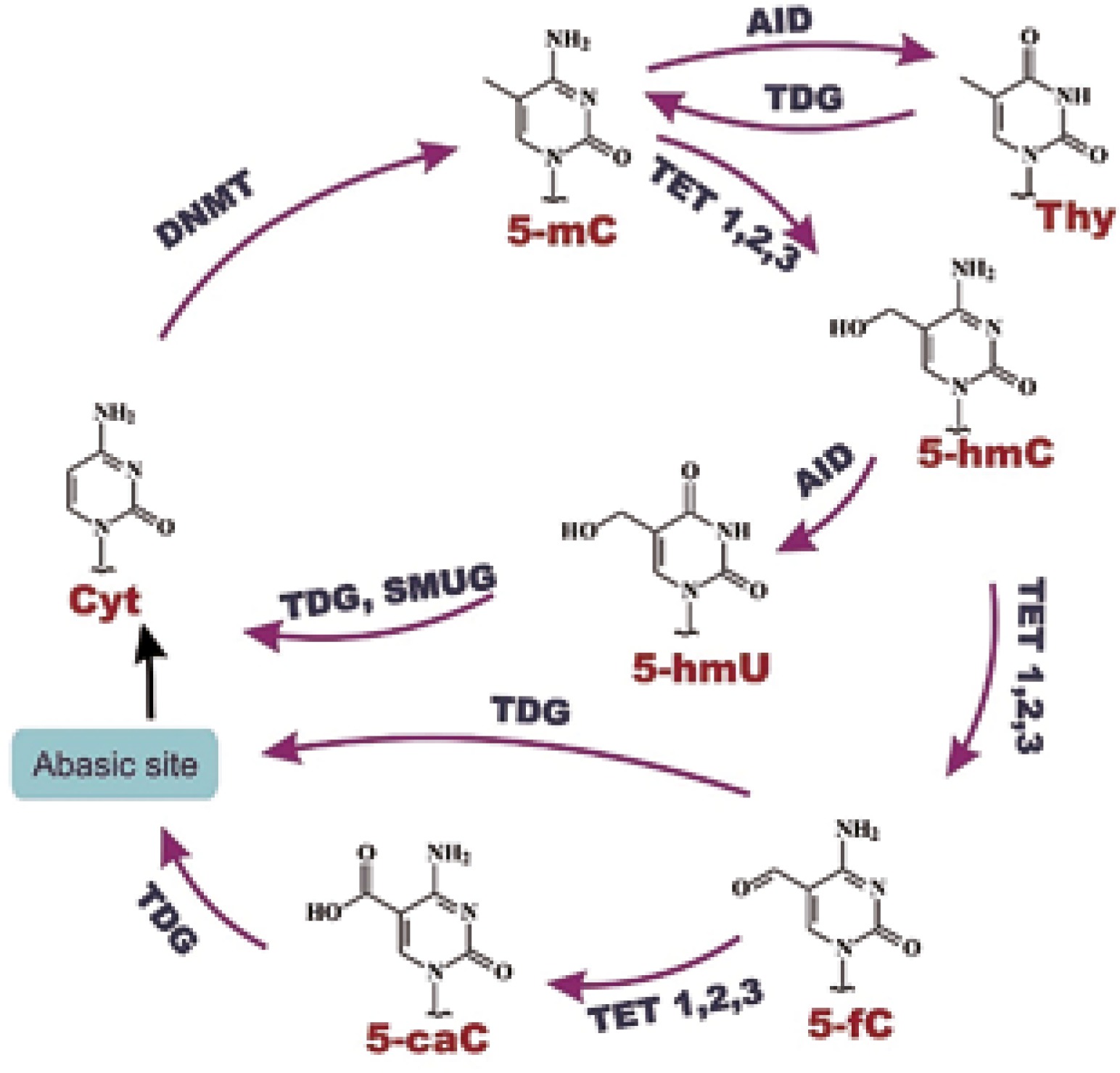
Epigenetic modifications cause disturbances in regulation of gene expression, including drug metabolism and distribution [1]. A considerable amount of literature has been published on the connection of 5-hmC with cancer development. It was found that many cancers reveal DNA hypomethylation [20–22]. A large and growing body of literature data has demonstrated that changes of 5-mC level imply 5-hmC depletion in many types of cancers [23, 24]. Literature data suggest that 5-hmC may be involved in gene regulation [25]. Moreover, an impaired DNA methylation pattern is linked with activation or repression of key genes, which in turn may lead to expression changes and cancer progression [7].
5-hmC level is regarded as a biomarker of malignant transformation, which is considered in evaluating the active DNA demethylation process. The potential role of active DNA demethylation derivatives as a novel predictive factor linked with hormonal therapy outcome has not been investigated yet. Recent research has revealed that 5-hmC is decreased in cancer cells, including breast cancer cells [23, 26]. Literature data suggest that 5-hmC level is positively associated with 5-mC level in breast cancer patients. Moreover, it was demonstrated that decreased levels of 5-mC and 5-hmC are potentially correlated with poor disease-free survival (DFS) and disease-specific survival (DSS) in breast cancer patients with ER/PR(-) in comparison to those with ER/PR(+) [27]. Although literature data indicate that 5-hmC is reduced in many types of cancer, there is a gap in terms of its possible predictive role in therapy outcome.
Recent developments have focused on the role of TETs and TDG in breast cancer patients. Yang et al. investigated the relationship between TET1 mRNA level and overall survival among breast cancer patients treated with anthracyclines. Additionally, they suggested that high TET3 and TDG expression might be a significant predictive factor of clinical outcome in this group [28]. However, it still remains unclear why TET and TDG expression is unsettled in different types of tumors. Since 5-hmC and its modifications are products of TET proteins’ activity, it is suggested that alterations in gene expression are associated with 5-mC derivatives content. Since the overwhelming majority of alterations in TET activity are not related to genetic mutations, it suggests that other factors are potentially responsible for such changes.
Breast cancer subtypes
Breast cancer is the most frequent malignancy amongst women worldwide. It affects over 2.1 million women per year globally and it is the cause of death for almost 600 thousand of them [29]. This type of cancer, similarly to others, displays global hypomethylation as a result of genome instability. Furthermore, it was conclusively demonstrated that alterations in DNA methylation of pivotal genes (BRCA, p53, ERα) are involved in cancer progression [30]. Changes in DNA methylation are associated with molecular subtypes of breast cancer, which may suggest an important role of impaired DNA methylation in carcinogenesis [31].
Breast cancer is classified into five biological subtypes based on expression of ER, PR, human epidermal growth factor 2 (HER2) receptors, and nuclear antigen Ki-67 (Table 1). According to the 5th St Gallen International Breast Cancer Conference, expression of steroid hormones and HER2 receptors constitutes a crucial prognostic and predictive factor of breast cancer outcome [33]. The vast majority of breast cancer cases (approximately 60–70%) are ER(+) and PR(+) positive. Breast cancers with high expression of steroid hormone receptors are less invasive, and have better prognosis overall. The triple negative subtype has the worst prognosis, and it accounts for approximately 15–25% of breast cancer cases. Regardless of potential clinical recovery, receptor expression determines the treatment strategy: trastuzumab for HER2 (+) subtypes or drugs affecting estrogen synthesis or estrogen receptors for those with ER/PR expression [32].
Table 1
Biological subtypes of breast cancer (based on [32])
Breast cancer receptors’ significance
ERs are members of the nuclear receptor superfamily of ligand-activated transcription factors. After ligand activation, ERs undergo conformational changes, and as a result they could bind to estrogen response elements (ERE) in DNA and have an impact on regulation of gene expression [34]. Isoforms of ER (ERα and ERβ) are under the control of two genes, ESR1 and ESR2. Contrary to ER, isoforms of progesterone receptor (PRA and PRB) are coded by one gene, PGR [35]. The expression and activity of PR are regulated by ER: PR is expressed as a result of ER activation [36].
The elevated estrogen activity in cancer cells is connected with increase of ERα quantity; thus ERα is used as a target of hormonal therapy of breast cancer. Moreover, the grade of malignancy and stage of differentiation are associated with ERα expression. In contrast to ERα, ERβ is expressed mainly in healthy mammary gland [37]. Moreover, ERβ could exert an antagonistic effect on ERα action in certain tissues, which in turn may lead to decrease of cellular proliferation. Reduced ERβ expression in cancer suggests that this isoform has suppressor activity in hormone-dependent tissue, e.g. in mammary gland [38].
In 2000 Filardo et al. observed that the rapid response to 17β-estradiol is a consequence of extracellular regulated kinase (ERK) activation, which was not connected with ERα or ERβ, but with a G-protein-coupled receptor named GPR30/GPER [39]. Later, it was conclusively demonstrated that GPER also binds estradiol with high affinity and is connected with rapid non-genomic signaling of estradiol [40]. GPERs are classified as membrane receptors, although they may also occur in cytoplasm and nucleus [41].
The HER family is arranged in regulation of growth and development in breast cancer cells. HERs, in contrast to ER and PR, are epidermal growth factor receptors (EGFR) expressed in the cell membrane. Due to the fact that HER2 acts without a known ligand, it constitutively occurs in active conformation, and undertakes dimer formation with another EGFR. Hetero- or homodimerization leads to tyrosine kinase phosphorylation, and activation of the signaling pathway [42]. HER2 (+) occurs only in 15% of breast cancer patients; however, 10% of them also expressed ER(+) [43].
It is becoming increasingly clear that there is a high probability that abnormal cell growth found in breast carcinoma might be the result of impaired up-regulation of ER, GPER and HER2. The potential signaling pathways are able to stimulate each other: G protein-coupled estrogen receptor can trigger HER2 signaling, while tyrosine kinases cascade preceded by HER2 activation may phosphorylate and initiate the activation of ER and its proteins [44, 45]. Receptors’ expression in breast cancer determines the clinical outcome. Hence, it could be possible that the DNA methylation pattern varies between human breast cancer cells with diversified expression of receptors.
Estrogens as natural ER ligands are implicated in growth and proliferation of cells, e.g. in mammary gland. Nevertheless, excessive estrogen exposure may have an impact on promotion and progression of breast cancer in humans [46]. Inhibited proliferation of cancer cells after high concentrations of β-estradiol (E2) was also observed in human cancer cell lines [47]. Moreover, E2 may act as a gene expression regulator though its ability to bind ER. Based on literature data, it was suggested that E2 can affect DNA methylation by promoting demethylation of CpG islands in promoter regions of genes [48, 49]. Furthermore, a recent study revealed that E2 supplementation of cultured cells resulted in almost entire removal of 5-mC in the SVEP1 gene promoter, and thus increased the unmethylated DNA level [50].
Biochemistry of tamoxifen
There are three possible treatment strategies of hormone-dependent breast cancer: arresting of estrogen synthesis via aromatase inhibitors, competitive binding to estrogen receptors and modulating their activity by antiestrogens, and prevention of ER signaling by causing degradation of ER by selective estrogen degraders (SERDs) [51]. The second group, represented widely as selective estrogen receptor modulators (SERMs), has been in clinical use for nearly 40 years [52].The best known representative of SERMs is tamoxifen, which is currently used in all stages of ER(+) breast cancer in pre- and postmenopausal women, and in ductal carcinoma in situ [53]. Moreover, tamoxifen is used for prevention of breast cancer for women at very high risk of developing the disease [54]. Additionally, longer tamoxifen treatment, up to 10 years, results in a 50% decrease in breast cancer mortality in the course of the second decade after diagnosis [55]. Based on pharmacological research, tamoxifen is transformed into three active metabolites in the human organism: 4-hydroxytamoxifen, N-desmethyltamoxifen, and 4-hydroxy-N-desmethyltamoxifen (endoxifen) [56]. Conversion of tamoxifen implicates hepatic CYP2D6 and CYP3A4/3A5 cytochromes, which metabolize tamoxifen to its metabolites in two different manners: to N-desmethyltamoxifen (CYP3A4/3A5), and further to 4-hydroxy-N-desmethyltamoxifen (CYP2D6), or to 4-hydroxytamoxifen (CYP2D6) and then to 4-hydroxy-N-desmethyltamoxifen (CYP3A4/3A5) [57]. It has been established by pharmacological profiling that tamoxifen is a prodrug, hence its therapeutic action results from its active metabolites: 4-hydroxytamoxifen and endoxifen [58]. The affinity of 4-hydroxytamoxifen and endoxifen to ERα and ERβ is considerably similar, much the same as the other antiestrogens associated with regulation of estrogen-dependent gene expression [59]. However, given that the plasma level of endoxifen is 5–10 fold higher compared to 4-hydroxytamoxifen, it is suggested that this metabolite is presumably a key tamoxifen derivative accountable for pharmacological activity of tamoxifen in the human organism [60].
Mechanisms of SERM action in breast cancer cells
SERMs may act as antagonists or agonists of ERs depending on type of receptor: nuclear or G protein-coupled. The antagonistic action of SERMs is associated with nuclear ERs and connected to breast cancer treatment. After binding to ER, tamoxifen evokes the receptor’s conformational changes, but distinct from the ER-estrogen complex. The tamoxifen-ER bond dimerizes and translocates to the nucleus, where it prevents transcription of estrogen-dependent genes by attaching to estrogen response elements (ERE) in DNA [61]. This successively results in inhibition of estrogen activity, which is connected with growth and proliferation of cells [62, 63]. As estrogen receptor agonists, SERMs affect GPERs, which has been identified as a main factor in rapid responses to estrogens [64]. Tamoxifen activation of GPER results in modification of the estrogen receptor or its co-activators by phosphorylation, which can cause independent ligand activation or an impaired response to other ER regulators [27] (Fig. 2). This type of interaction occurs mainly in bones and liver [3]. However, it is possible that estrogens and SERMs affect impaired cell growth and proliferation though activation of GPERs, and it may be the predominant pathway in transformation of breast cancer cells into cells resistant to hormone therapy. Hence, it could conceivably be suggested that the role of tamoxifen is not only limited to involvement in regulation of estrogen-induced genes.
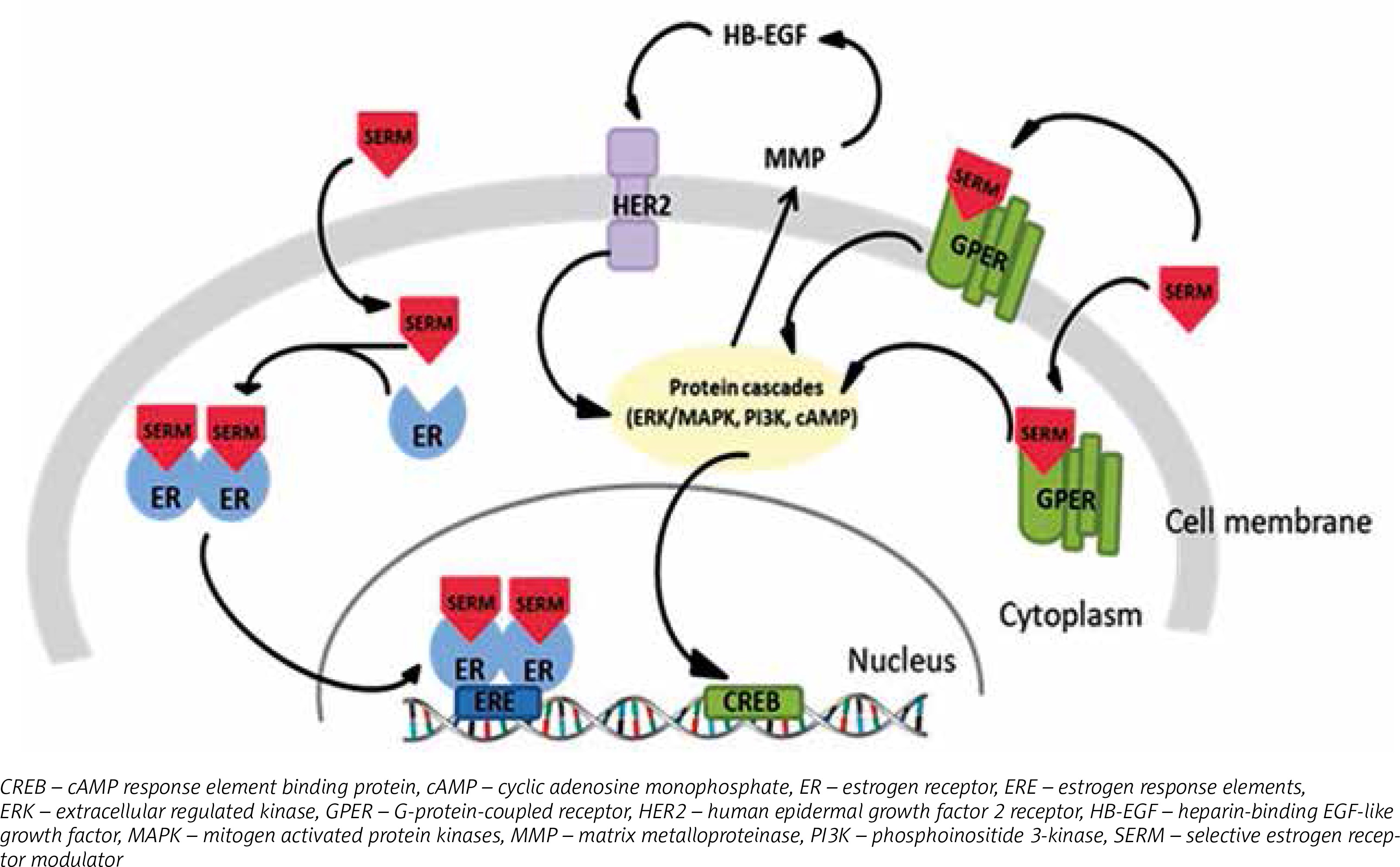
Tamoxifen as an antiestrogen may inhibit breast cancer cell growth and proliferation. However, acquired resistance to SERMs therapy still remains a problematical issue as adjuvant therapy is extended. The proposed mechanism of antiestrogen resistance is associated with GPER activation. It was suggested that GPERs may precede stimulatory estrogen signaling for proliferation and migration, regardless of ER expression [65]. Moreover, abnormal activity of ER caused by antiestrogens may be linked with changes in gene expression as a result of epigenetic modifications [8].
It was conclusively demonstrated that estrogen receptor signaling and DNA methylation are implicated in regulation of the cell cycle by modulating antiproliferative and proliferative genes [66, 67]. Although there are some data concerning partially overlapping gene regulation by estrogen signaling and DNA methylation, its detailed molecular mechanism and common target genes have remained enigmatic [8, 68]. It may be possible that SERMs can potentially interrupt estrogen receptor function and thus similarly affect the DNA methylation pattern. Moreover, several studies have proved that sustained SERM therapy can manifest distinct global gene expression and DNA methylation of promoters in breast cancer cells [69, 70].
Conclusions
The treatment of choice of ER/PR(+) breast cancer patients is based on the selective estrogen receptor modulator tamoxifen, which in long-term therapy serves as a considerably reliable drug. Of note, tamoxifen therapy leads to recovery in hundreds of thousands of ER/PR(+) breast cancer cases [71]. Since the discovery of tamoxifen, the use of the drug has evolved by embracing its ability to act like estrogen or antiestrogen depending on the receptor type and its localization around the body. Such ability created new possibilities in drug development and therapeutic use, i.e. tamoxifen acts like estrogen in osteoporosis treatment by preventing bone density loss, and like antiestrogen in breast cancer therapy by inhibition of estrogen action [72]. Hence, it is becoming increasingly clear that the action of SERMs at different target sites is more complex than just switching estrogen activity. It may be dependent on other factors and processes taking place in target cells.
Further studies are needed to determine whether tamoxifen, similarly to steroids, may evoke changes in the methylation pattern. Such research may provide new information, which may pave the way for new diagnostic and therapeutic methods as well as innovations in personalized medicine approaches.






