
Introduction
Anaplastic thyroid cancer (ATC) represents by far the most aggressive type of primary malignant thyroid cancer, arising from transformation of differentiated thyroid carcinoma or de novo and constituting 1–2% of all thyroid malignancies, but having the greatest clinical impact on cancer-related deaths [1, 2]. In Europe, the cancer incidences, given as age-standardized rates, are low at < 0.3/100,000 in females and < 0.2/100,000 in males [3]. Findings from an epidemiological study in the United States indicated that the incidence rate from 1973 to 2014 increased by 3.0% annually [4]. In Europe, however, cancer incidence rates appear lower with an annual percentage change (APC) of +1.3% according to a large Dutch study [5], and even decreasing rates in females and males were noted according to a large Danish study [6]. Anaplastic thyroid cancer mostly affects older patients, with the majority being over 60 years old, and it is commonly diagnosed at an advanced stage of disease (by definition stage IV according to the staging system of the American Joint Committee on Cancer) with rapid tumour progression and local or distant metastases in up to 30–40% of patients [7, 8]. The diagnosis of ATC requires thorough conventional and immunohistochemical exclusion of differential diagnoses such as well-differentiated thyroid carcinoma (WDTC), poorly differentiated thyroid carcinoma (PDTC), medullary thyroid carcinoma (MTC), angiosarcoma, sarcoma, lymphoma, squamous cell carcinoma, metastasis to the thyroid from solid tumours, and Riedel thyroiditis. The concomitant resistance to radiotherapy and systemic chemotherapy contributes to extremely poor overall survival (median 3–10 months; overall disease-specific mortality rate: 68.4% at 6 months, 80.7% at 12 months) despite multimodal aggressive first-line therapeutic approaches [7–10]. With increasing knowledge of the tumour biology, the identification of the underlying genetic pathways, modifications of the transcriptome and proteome and associated immunomodulatory mechanisms of ATC on the one hand, and the emerging role of novel, molecular-based single/multi-targeted therapies on the other hand, a growing number of human early clinical trials can be noted [11, 12] (www.clinicaltrials.gov). In addition, international and national guidelines need to be adapted and brought up to date to evaluate established procedures and to implement evidence-based findings, because the recommendations of the American Thyroid Association date back to 2012, those from the National Comprehensive Cancer Network to 2017, and from the British Thyroid Association to 2014. There are currently no available standardized European or German treatment guidelines (German S3-Leitlinie, registration number 031-056OL, is scheduled for 31 December 2021 according to Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e. V).
In recent years, several studies have confirmed that the molecular profile of ATCs includes a small proportion of tumours that demonstrate acquired mutations in the DNA mismatch repair (MMR) pathway (up to 10–15%), being linked to impaired or deficient post-replicative DNA repair mechanisms [13–19]. As a functional consequence, errors that occur during replication processes might not be recognized properly and be corrected, leading to a dysregulation of MMR and resulting in length aberrations of short repetitive DNA sequences (called short tandem repeats [STRs] or simple sequence repeats [SSRs]) along the whole genome. This results in microsatellite instability (MSI), which is known to drive carcinogenesis and tumour progression [14, 17, 20].
Therefore, this data analysis aimed to systematically review and meta-analyse the prevalence of impaired DNA MMR status or MSI in ATC and investigate its potential prognostic/predictive value in view of available targeted therapies, with the intention of ensuring optimal individual patient care.
Material and methods
Formal consent was not required for this review.
Search strategies
The search was conducted in different electronic databases (PubMed [MEDLINE], Google Scholar, and ASCO), and libraries of interest were imported into the Mendeley citation manager. Search terms included “anaplastic thyroid carcinoma”, “anaplastic thyroid cancer”, “DNA mismatch repair”, “mismatch repair deficiency”, and “microsatellite instability”. Search filters were applied as follows: text availability (abstract and full text), species (human), language (English), and source type (journal). This investigation followed the Preferred Reporting Items for Systematic Review and Meta-analysis (PRISMA) [21].
Critical assessment of selected articles and data acquisition
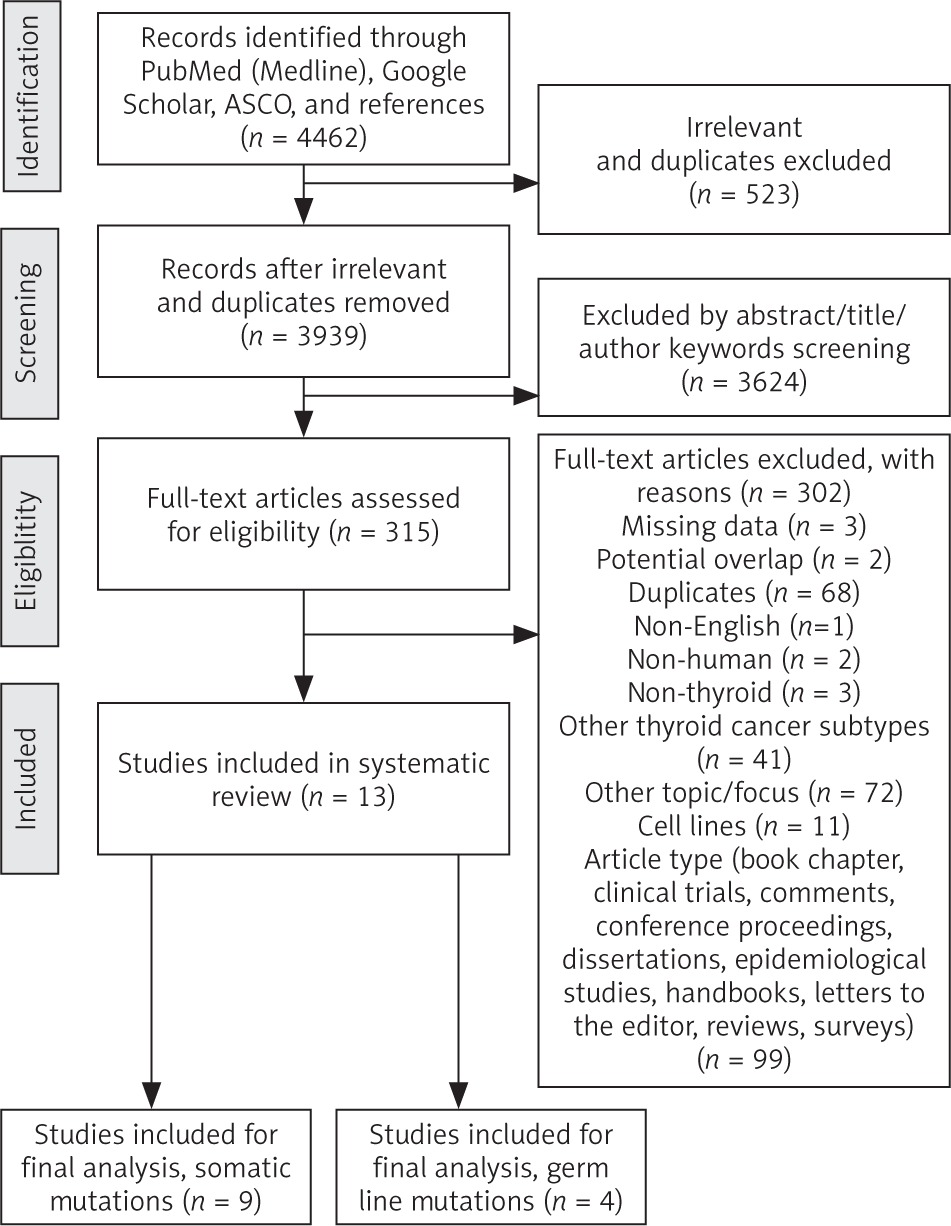
Research for appropriate data followed standardized predefined parameters. Title, abstract, and author keywords were screened for all recorded studies, and 2 authors (MLR and PC), each a consultant of pathology, independently reviewed and critically discussed the selected data. The quality if the studies and their potential bias were evaluated. The process of categorization into subgroups and final study selection was performed as shown in the PRISMA flowchart, adapted and modified in accordance with Moher [21] (Fig. 1). Extracted information included: first author, year of publication, total case number, number of included and analysed ATCs, detection methods, and MSI rate. Additional information, such as molecular phenotype, detected mutations in the MMR genes, type of molecular modifications in the MMR genes, frequent co-mutations, prior treatment, and clinical features, was not considered as mandatory, but optional. Studies with potential overlap, an insufficient reference panel, or isolated MMR gene investigation were excluded from the final analysis.
Results
Results from systematic data research and meta-analysis
A systematic search approach in different electronic databases initially identified 4462 citations, from which 315 potential full-text articles were finally checked for eligible criteria. Of these, 13 studies qualified for final analysis, 9 of them considering somatic mutations, and 4 of them documenting germ line mutations in the setting of Lynch syndrome (Fig. 1). The inclusion criteria and additional information of the 9 studies without suggested hereditary association are listed in Table 1 and served for meta-analytic evaluation. In total, 175 cases diagnosed with ATC were tested for MSI. Among them, 13 cases (7.4%) were verified with MSI+ status by different detection methods. For 12 of these cases, the molecular phenotype was given, with 11 cases (91.6%) presenting with a disproportionally higher tumour mutational burden (TMB; ‘hypermutated’ phenotype) as a genetic signature (Table 1, Fig. 2) concordant with previous reports in the literature [13–18, 20, 22]. In the landscape of ATC, the affected and documented MMR genes thus far include MutL homologue 1 (MLH1), MutS homologue 2 (MSH2), MutL homologue 3 (MLH3), MutS homologue 5 (MSH5), and MutS homologue 6 (MSH6) (Table 1), with mutations in the MSH2 gene (33%) being the most frequent, followed by MSH6 (25%) and MLH1 (16.7%) (Fig. 2). Mutations in 2 MMR genes occurred in combinations as follows: MLH1-MSH2 (8.3%), MSH2-MSH6 (8.3%), and MLH3-MSH5 (8.3%) (Table 1, Fig. 2). Interestingly, mutations in the gene encoding the MMR endonuclease PMS2 have hitherto not been described, which could be suggestive of low frequencies of PMS2 mutations in general or indicative of more complex underlying mechanisms, implying insufficient detection methods or difficult data interpretation so far. A study conducted on patients with colorectal cancer (CRC) in search for germline mutations in PMS2 (after having found an isolated loss in the immunohistochaemia but preserved MLH1 expression) described significant complications in the detection of mutations due to paralogous genes [23]. Final analysis additionally revealed 66 co-mutations documented in 9 cases, with TP53 (88.9%), NF1 (44.4 %), ATM (33.3%), and RB1 (33.3%) being the most commonly detected (Fig. 3). These were followed by APC, ASXL1, BRAF, DAXX, KMT2A,MAP3K1,MEN1,MTOR, PIK3CA,PTEN,SMARCB1, TERT, and TSHR (each 22.2%) (Fig. 3). Of note, RAS mutations have not been reported. Survival ranged between 2.8 and 48 months (n = 9), and patient age varied between 49 and 84 years (Table 1).
Fig. 2
Distribution of mutated MMR genes within the MSI-positive ATC cohort of the finally included studies (available for n = 12) and the proportion of hypermutated phenotype status (available for n = 11)
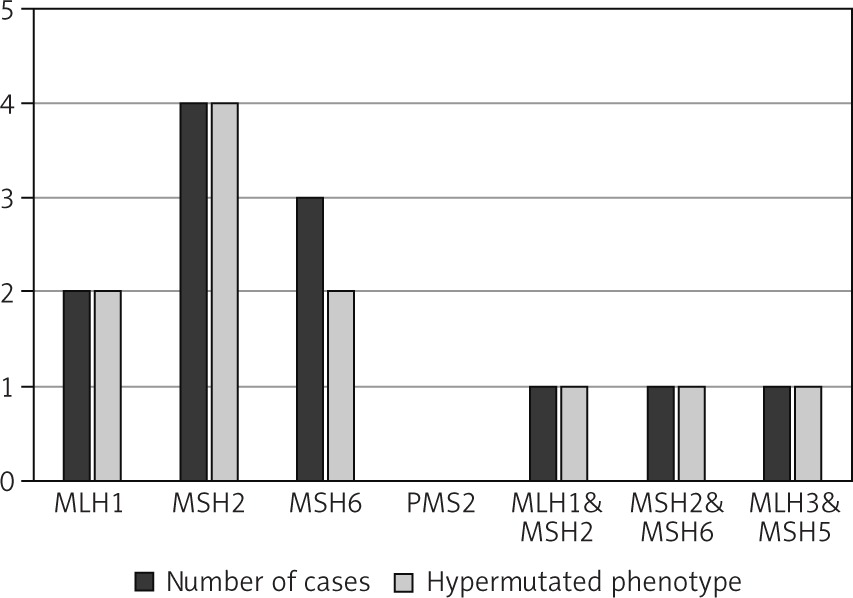
Fig. 3
Distribution of co-mutations within the MSI-positive ATC cohort of the finally included studies (available for n = 9)
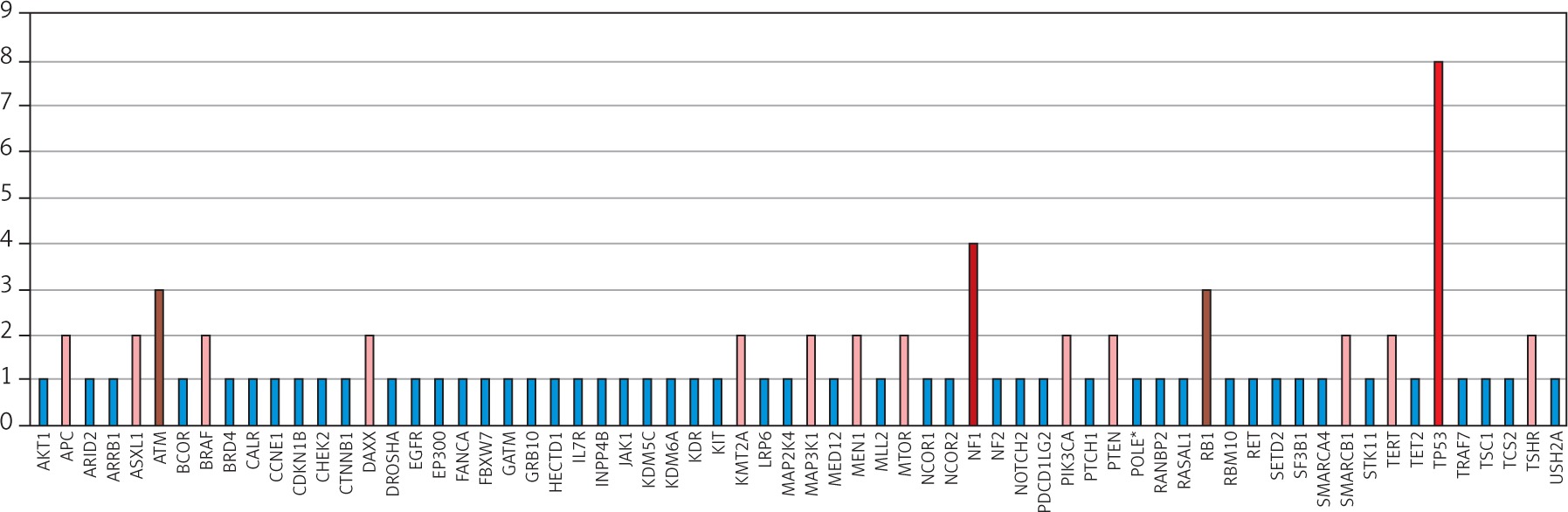
Table 1
Summary and characteristics of included original references with comment on DNA mismatch-repair status in anaplastic thyroid cancer
[i] ADR – adrenal, ATC – anaplastic thyroid cancer, F – female, HTN – hypertension, IDC – ductal adeno-carcinoma of the breast, M – male, MMR – DNA mismatch repair, mos – months, NA – not applicable, ND – not detected, NGS – next-generation sequencing, OSS – osseous, OTH – other, PCR – polymerase chain reaction, PMR – polymyalgia rheumatic, PUL – pulmonal, qRT-PCR – quantitative real-time PCR, T2DM – type 2 diabetes mellitus, UC – ulcerative colitis, WES – whole-exome sequencing, WGS – whole-genome sequencing, XBRT – external beam radiotherapy, y – years, a – non-radioactive external PCR and radioactive internal (nested) PCR, β – one case with MSH2 and MSH6 mutation, c – mutations not specified for each MSI case, d – one additional case with MMR-d, but MSS/borderline MSI status was excluded, e – no BRAF or RAS mutation detected, f – borderline MSI+, γ – no disease-specific neoadjuvant therapy
Remarks on included studies
The study of Wong et al. demonstrated, in a series of 28 ATCs, that MMR-deficient (MMR-d) and MMR-proficient (MMR-p; intact MMR protein expression) tumours did not show any dissimilarities in clinicopathological parameters such as macroscopic tumour size, extrathyroidal tumour expansion, positive nodal status, resection margin status, or histological pattern [16, 24]. Interestingly, however, 3 cases showed a prominent perilesional inflammatory infiltration. The prevalence of MMR-d status was given with 4/28 ATCs (14%), although 1 case could not convincingly be verified as MSI lacking mutations in the DNA MMR genes MLH1, MSH2, MSH6, PMS2, and EPCAM. It was also reported by Wong that all cases were devoid of WDTC, only partially presenting with foci of PDTC [16, 24]. Nevertheless, MMR-d cases tended to have significantly better overall survival rates (1 case reaching 48 months) and presented with fewer metastases at the time of diagnosis [16]. The study of Ravi also included 1 case with borderline MSI status [18]. For only 1 case, an associated well-differentiated tumour component was reported [14]. Three of the studies did not detect MSI cases [25–27].
Comment on excluded studies
Potential overlap was identified in 2 studies [13, 17], which were therefore excluded. A panel of only 3 microsatellite markers with isolated focus on MSH2 was also not considered in the final analysis [28]. The study by Dong et al. was not taken into account either, because the discovered MSH6 mutation in 1/5 ATCs was only detected in the well-differentiated tumour component (PTC) [29]. Potential risks of bias were identified during data extraction and include neoadjuvant therapy, differences in applied molecular testing methods, and possible ATCs as a minor component only. The latter fact was not specified by any study included.
Original case reports with anaplastic thyroid cancer manifestation within the spectrum of Lynch syndrome
On extremely rare occasions, ATC (and PTC) may manifest as a non-typical index tumour within the spectrum of Lynch syndrome (LS), an autosomal dominant inherited cancer syndrome, formerly known as hereditary non-polyposis colorectal cancer syndrome. Carriers of germline mutations in the DNA MMR genes have a significantly increased risk of early cancer development, characteristically of the colon/rectum, endometrial, skin, ovaries, kidneys, ureter, bladder, stomach, intestinal, pancreatobiliary system, and brain. In general, approximately 90% of DNA aberrations are found in the genes MLH1 and MSH2, whereas barely 10% are noted in the genes MSH6 and PMS2 [30]. Interestingly, with regard to ATC, only 3 cases with documented germline mutations in the DNA MMR genes MSH2 and MSH6 have been reported so far [31–34] (Table 2). Of note, the 2 cases with MSH2 mutations presented with MSI-L status (in thyroid), whereas the MSI status for the case with MSH6 mutation was not given in the 2 published reports, but a hypermutated phenotype was protocolled (Table 2).
Table 2
Summary and characteristics of included original case reports with anaplastic thyroid cancer manifestation within the spectrum of Lynch syndrome
Differences in testing panels
In a larger scope, a review of the literature revealed a noteworthy study analysing MSI status with an adjusted reference panel, with special emphasis on thyroid cancer-specific loci (including microsatellites in the genes THRA1, thyroid hormone receptor α, and TSHR, thyroid-stimulating hormone receptor; RET, p53), showing interesting clinicopathologic correlations: (I) MSI levels were as follows: THRA1 (36.5%) > D2S123 (32.4%) > D11S912 (27.6%) > D2S115 (22.4%) > RET (18.8%) > TSHR (18.4%) > p53 (17.1%) > D2S399 (14.3%) > BAT-26 (8.3%); (II) MSI within the TSHR gene correlated with older patient age (> 70 years) and metastases to regional lymph nodes; (III) MSI in the dinucleotide marker D2S123 was detected in all FTCs (100%), but only in a quarter of non-FTCs (25.3%); and (IV) the mononucleotide marker BAT-26 displayed the lowest frequencies [35]. In contrast, the study by Genutis et al., including more than 480 tumour probes of different cancer types (PTC [n = 196], FTC [n = 156], PDTC [n = 18], ATC [n = 51], MTC [n = 65] and oncocytic carcinoma [formerly Hürthle cell thyroid carcinoma; n = 3]), analysed MSI status based on PCR and NGS, applying the Bethesda panel (BAT-25, BAT-26, D2S123, D17S250, D5S346). Remarkably, MSI was only detected in 4/156 FTCs (2.5%) and was absent in all remaining 327 cases with other histomorphologies [25]. Of the 35 FTCs investigated by PCR herein, 2 cases presented instability at multiple microsatellites (case 1: BAT-26, D2S123; and case 2: BAT-25, BAT-26, D17S250) [25]. In this context, a third study should be mentioned: Santos et al. analysed 96 thyroid probes including PTCs (70 cases), FTCs (12 cases), follicular adenoma (FA, 7 cases), and normal tissue (7 cases) based on PCR, using the Bethesda panel plus 2 additional markers: BAT-40 and D11S912. The following astonishing frequencies of MSI status were given: 59/70 PTCs (84%; MSI-H 64%, MSI-L 36%), FTCs 11/12 (92%; MSI-H 82%, MSI-L 18%), and 6/14 FAs (43%; MSI-L 100%). MSI levels were as follows: D17S250 (37%) > D5S346 (34%) > D2S123 (19%) > D11S912 (12%) > BAT-40 (10%) > BAT-26 (10%) > BAT-25 (2%) [36]. These data also illustrate higher MSI frequencies in follicular-derived lesions. Comparable data were presented by Mitmaker et al. with MSI detected in 9/14 PTCs (64%), and 10/16 FTCs (62.5%), whereas 9/10 FAs were microsatellite-stable (MSS) or MSI-L [37]. However, PDTC and ATC cases were not the subject of the investigations of the latter 2 studies.
Discussion
Taking the facts of these studies together and earlier recommendations by the National Cancer Institute (NCI) into account, the detection of MSI in thyroid cancer and across other malignancies might require an adapted next-generation sequencing (NGS) reference panel and choice of different marker type in distinct histological subtypes [38]. This valuable approach is supported by data presented by Hause et al., who in an exome-based study investigated more than 200,000 microsatellite loci in 18 different cancer types (n = 5930 cases) (including PTC) and found cancer-specific loci that were more suitable in MSI detection [39]. The data of the studies listed above also indicate that a mononucleotide marker may not be appropriate in the setting of thyroid neoplasms [35–37], thus differing from its value in CRC and endometrial cancer (EC). Against this background, the major differences in the detection frequencies of MMR-d/MSI in variable thyroid neoplasms can be explained, with some studies illustrating a complete absence [28, 40]. Differences in detected prevalences might also result from varying molecular testing methods (e.g. targeted DNA sequencing [NGS], whole-exome sequencing [WES], whole-genome sequencing [WGS], polymerase chain reaction [PCR], capillary electrophoresis, and RNA-sequencing), specific sequencing platforms, and defined cut-offs (MSI-L versus MSI-H, number of tested microsatellite markers). Of note, optimal DNA sequencing coverage (or depth) is desirable, and better results have been achieved by NGS (IMPACT) compared to WES [15]. Beyond that, several computational tools have been introduced to screen the generated sequencing data (from NGS, WES, WGS) for MSI (e.g. MANTIS, mSINGS, MSISensor, MonoSeq, MOSAIC) [25, 39, 40], which may contribute to data standardization and identification of new MSI signatures in many cancer types.
Because the exact role of MSI in thyroid cancer initiation and dedifferentiation processes is still not well understood, there is controversy as to whether MMR-d/MSI represents a feature of early or late-stage disease. This can be explained by the fact that DNA MMR events or MSI are not only found in the later stages of PDTC and ATC, but also in the preceding FTC and PTC lesions, as well as in benign and tumour-like lesions such as FA and nodular hyperplasia/nodular goitre [14–17, 19, 22, 25, 35, 41–44]. Soares and Lazzereschi even documented 2 benign lesions with MSI-H status (1/13 nodular goitres plus 1/15 FAs and 2/16 FAs, respectively) [19, 41]. Conversely, Hause et al. interpreted and discussed their MSI data along a continuum with MSI contributing to cancer progression [39]. This interpretation had already been made by Lazzereschi et al., who had found MSI (+) in lymph node metastases in 2 MSI (–) thyroid carcinomas (1 PTC, 1 HTC) and documented an increased prevalence of MSI (+) status at higher clinical stage, suggesting that MSI contributes to tumour expansion and evolution [19]. In line with this, an elegant single-case study by Paulsson et al. illustrated the coexistence of FTC, PDTC, and ATC, which were shown to be derived from 1 clone and presented an MSI signature in the PDTC and ATC parts, whereas FTC had borderline-MSI status [22]. Clonal evolution studies of Dong et al. led to the proposal of 2 ways of ATC progression: 2/5 ATCs with associated WDTC showed shared subclones, and 3/5 ATCs with associated WDTC had an independent clonal architecture [45].
Moreover, there are still insufficient knowledge and data concerning the actual prognostic value of MMR-d/MSI status with inconsistent, in part contradictory, analyses. In a large study conducted by Xu et al., including 360 ATCs, the aforementioned better prognostic outcome [16, 35] could not be reproduced [13]. Of note, the study by Onda et al. included only 2 ATC cases within a heterogenous cohort predominated by PTCs (85.6%) [35]. A positive (nonsignificant) relationship between MSI+ and prognosis was also reported by Hause et al., but the included TC cohort was not further specified as to histological subtypes [39]. An associated poor prognostic outcome of MSI+ tumours was discussed by Soares and Mitmaker, with both studies having exclusively analysed WDTCs (FTC, PTC) along with benign lesions, whilst PDTC and ATC cases were not the subject of the studies [37, 41].
Accordingly, these data cannot provide reliable conclusions or predictions because the prognostic impact of MMR-d/MSI may differ in each histological subtype and stage of disease. Differentiated assessment of the prognostic relevance of MMR-d/MSI was already given in other malignancies (e.g. CRC, EC, gastric cancer, oesophageal cancer) [46–49], and staging parameters were shown to be relevant in part [50, 51]. Future research and clinical investigations should also take prior treatments (chemotherapy, radiotherapy, etc.) into account because study results of other tumours, especially CRC and gastric cancer, tended to show a distinct association between MMR-d/MSI status and chemotherapy response [46, 49, 51]. It further remains an open issue as to whether the association or increased frequencies of MMR-d/MSI in lesions with a follicular or follicular-derived morphology, as described by a few studies, represent robust results [22, 25, 35, 36].
Particularly noteworthy are the frequent co-mutations in the tumour suppressor gene TP53 in the setting of MMR gene modifications [14, 15, 17, 22]. Various genes of the DNA repair system display p53-corresponding sites, and it is suggested that the transcription factor p53 contributes to the transcriptional regulation of these genes, including MMR members such as MSH2 [22, 52]. Combined immunohistochemical studies of MMR/p53 might be of use and need to be explored because distinct expression patterns have been described in other tumour locations [53–55].
Besides somatic mutations in the MMR genes, inactivation of these repair mechanisms may also be caused by epigenetic modifications. In particular, promoter hypermethylation was reported for the DNA MMR gene MLH1. In a study by Guan et al., the prevalence of MLH1 methylation in 38 PTCs came to 21% (8/38 PTCs), and an association with the BRAF V600E mutation as well as a positive nodal status was shown [56], thus demonstrating parallels to CRC [57, 58]. In the study by Santos et al., MLH1 methylation frequencies were given as follows: 44% PTCs (31/70 cases), 33% FTCs (4/12 cases), and 64% FAs (9/14 cases), and only a slight (“marginal”) association with MSI status was postulated for the PTC cohort. However, no association with the BRAF V600E mutation was found [36]. Because there are currently very few data available in the area of FTC, PDTC, and ATC, further research and analyses are required. The same applies to MSH2 promotor hypermethylation as a result of EPCAM mutations, being reported in CRC [59].
The prior immunohistochemical evaluation of antibodies recognizing the 4 DNA MMR proteins MLH1, MSH2, MSH6, and PMS2, which function as heterodimers (e.g. MLH1-PMS2, syn. MutLa; MSH2-MSH6, syn. MutSa), may increasingly be of diagnostic value. At present, though, there are insufficient data available in thyroid cancer in general and in ATC in particular with respect to its correlation with NGS findings or interpretation of aberrant (weak, heterogenous, discordant) staining patterns, as reported in other tumours [60–63]. Further research efforts are required to evaluate the immunohistochemistry of MMR proteins as a first-line screening tool. Because MMR-d tumours of other locations and origins are shown to potentially respond to novel immunotherapies and may be associated with different prognostic outcome, corresponding patients need to be identified and risk-stratified, and emerging therapeutic options should be explored in the setting of aggressive ATC.
Biomarker and target therapies for anaplastic thyroid cancer: limitations and perspective
A comprehensive and precise understanding of the genetic and proteomic landscape underlying ATC in general and thorough histomorphological, immunohistochemical, and molecular analysis in individual cases are crucial for the identification and implementation of new anticancer treatment strategies. To explain all current approaches in detail would go beyond the framework of this review. However, a few interesting aspects and developments shall be mentioned.
A variety of single/combined-targeted therapies already exist or are evaluated within phase II clinical trials (compare ClinicalTrials.gov). These include tyrosine kinase inhibitors (TKIs), such as pazopanib, multikinase inhibitors (MKIs), such as sorafenib, sunitinib, and lenvatinib, and selective BRAF kinase inhibitors, such as vemurafenib, dabrafenib, or encorafenib combined with the MEK inhibitors such as trametinib, cobimetinib, binimetinib, or selumetinib [11, 12, 64–68]. The United States Food and Drug Administration (FDA) has already approved combined therapy with BRAF (dabrafenib)/MEK (trametinib) inhibitor in ATCs carrying BRAF V600-mutations [11, 69]. Moreover, combined therapies, e.g. fosbretabulin with paclitaxel/carboplatin or dual pathway blockade with MAPK and PI3K/mTOR, have been evaluated, the latter with promising results in a single case study [70, 71]. One patient with a temporary response (18 months) to the mTOR inhibitor everolimus was also noted in a phase II study [72]. Moreover, the results of studies evaluating the NTRK inhibitors larotrectinib and entrectinib were presented lately, with a substantial tumour response [73–75]. This has already led to FDA and the European Medicines Agency (EMA) approval for solid tumours with NTRK gene fusions. Furthermore, selpercatinib, a selective inhibitor of RET kinase, has been approved for RET fusion-positive thyroid cancer [76].
Over the past years, immuno-oncologic treatments, especially immune checkpoint inhibitors (ICIs) (e.g. anti-programmed cell death-1 [PD-1], anti-programmed cell death-ligand 1 [PD-L1], and anti-cytotoxic T-lymphocyte-associated protein 4 [CTLA-4]) have revolutionized the field of anti-cancer therapies in many entities such as melanoma, non-small-cell lung carcinoma, Merkel cell carcinoma, renal cell carcinoma, classical Hodgkin lymphoma, squamous cell carcinoma of the head/neck, and urothelial carcinoma [77, 78]. These drugs apply to signal-transducing cascades, which modulate immune system responses, with the PD-1/PD-L1 axis downregulating T cell-mediated innate and adaptive immune responses and preserving immune tolerance via inhibiting feedback mechanisms in the Ras-Raf-MEK-ERK and PI3K-AKT pathways [79]. Given the capacity of some tumour cells and immune cells (especially CD8+ T cells) of the cancer-specific microenvironment to express PD-L1, the PD-1/PD-L1 pathway represents an effective immunoediting and tumour escape mechanism by suppressing the effector phase of T-cell activation [80, 81].
Interestingly, some ATCs demonstrate relevant upregulation of inhibiting immune checkpoint regulators. Recent studies found the expression of the PD-L1 on cancer cells in up to 20–30% of analysed ATCs and slightly over 10% of PD-L1-positive intratumoural immune cells [82, 83]. Another study by Cantara et al. (including 20 ATC patients) revealed more frequent expression of PD-L1 on tumour cells (70–90% of ATCs), additionally showing tumour regression in a representative mouse model after anti-PD-L1 antibody application [84]. Also, in a murine ATC model, the combined therapy of anti-PD-L1 and a BRAF inhibitor was even more successful in tumour size reduction and increasing the number of tumour infiltrating immune cells [85]. Of note, a high PD-L1 amplification level in ATC was detected in about 5% within a large series of solid tumours including 177 cases of ATC [86]. The status of immune checkpoint blockade – as a single agent or in combination therapies with BRAF/MEK inhibitors – in ATCs/metastasized ATCs is presently the subject of more than 10 clinical phase I/II trials (as of March 2021), including the anti-PD-1/PD-L1 antibodies pembrolizumab, nivolumab, atezolizumab, durvalumab, cemiplimab, and PDR001, among others (https://clinicaltrials.gov) [9, 10]. These trials were preceded by some very promising studies and single case reports that confirmed a favourable response, with one MMR-p case even demonstrating a complete response [82, 84, 87–89]. Encouraging results were also presented just recently after combined therapy with lenvatinib and pembrolizumab [90].
Nevertheless, the robustness of the immunopredictive value of PD-L1 to the immune checkpoint treatment response – as a solitary pan-tumour biomarker – has been questioned by several studies, in which this association could not be confirmed constantly [81, 91]. A synoptic report of different clinical trials analysing anti-PD1/PD-L1 therapy, including 1400 patients with solid tumours of varying entities, came to an overall response rate (ORR) of 48% in PD-L (+) tumours and, surprisingly, 15% in PD-L1 (–) tumours [92], making the necessity of a more reliable predictive biomarker clear. Accompanying difficulties arise from the challenging assessment of the PD-L1 immunohistochemistry, with distinct antibody applications for different tumour entities, variable cut-offs, defined scores, and interobserver variability in evaluating a true positive immunohistochemical reaction [93, 94], making meta-analytic research even more difficult.
The status of MMR deficiency or MSI was shown to be a further candidate serving as a predictive biomarker as regards the response to immune checkpoint blockade in several cancer subtypes [44, 95]. This is currently explained by the potential and ongoing generation of tumour-specific neoantigens (syn. mutation-associated neoantigens, MANAs) that results from high tumour mutational load, thereby inducing an immune response [44, 95–97]. With the extended approval of the immune checkpoint agent pembrolizumab (KEYTRUDA), a PD-1 receptor inhibitor, by the FDA in 2017 for all advanced staged, unresectable, or metastatic solid cancers in adult (and paediatric) patients with MMR-d or MSI-H status, independent of the tumour entity and primary tumour site (tissue/site-agnostic), oncologic treatment strategies and options have undergone a paradigm shift, and molecular testing of MMR-d/MSI has become increasingly important. The EMA, however, did not follow these recommendations. Instead, the tumour-specific immunopredictive responses were required thereupon to be evaluated precisely for each entity to receive treatment authorization, and in the meantime some progress has been made in various cancer types [44, 95, 98, 99].
Nevertheless, the expression pattern of PD-1/PD-L1 within this special molecular subtype of MMR-d/MSI signature in ATC is currently unknown. Further investigations are necessary to elucidate the impact of the PD-1/PD-L1 axis blockade in this distinct subgroup of ATC: (I) with respect to MMR-p +/– TMB high, (II) in the context of MMR-d/MSI-H plus TMB high, (III) in MSI-L tumours, (IV) in MMR-d/MSI-H without TMB high, and (V) in MMR-d/MSI-H without PD-1/PD-L1 expression but with a potential response, to name a few issues. The development of tumour type-specific, risk-stratified testing guidelines is also recommended.
It is finally worth mentioning that other potentially predictive biomarkers of response to ICIs in thyroid (and non-thyroid) malignancies are currently being discussed and include TMB, APOBEC, MANAs, IFNg, TILs, and driver mutations, among others [100, 101]. Addressable structures and future therapies also include TAMs and cancer vaccines [Ma 2020].
Conclusions
This study emphasized the small subgroup of ATCs with DNA MMR deficiency or MSI status. This subgroup obviously differs from conventional ATCs in a few clinicopathological parameters and is, in part, suggested to have better outcomes, although the tested cohorts were heterogenous and the prognostic value was not always applicable to ATC. However, given some promising results considering the response to immune checkpoint inhibitors in MMR-d/MSI-H tumours of other locations, ATCs also need to be analysed with a focus on the MSI signature, to identify patients with a distinct predictive/prognostic outcome, which can lead to adequate therapy. The results of immunotherapies in ATC, independent of MMR-d/MSI status, are currently being evaluated in numerous clinical trials, giving rise to the hope that improved treatment regimens will become available soon.
The diagnosis of ATC necessitates thorough differential diagnosis from other thyroid and non-thyroid malignancies as well as metastases to the thyroid. Tumour molecular profiling is fundamental in each ATC, not only for predictive and prognostic reasons, but also to elicit potential therapeutic options. Analysis of MSI status is strongly suggested and PD-L1 immunohistochemistry should be performed.






